
Abstract
Cisplatin is widely used as the standard gastric cancer treatment, but the relapse and metastasis are common as intrinsic or acquired drug resistance. CD133 has been widely known to be associated with chemoresistance in various cancer cells. In this study, we focused on investigating the function and mechanism of CD133 underlying cisplatin resistance in gastric cancer cell line KATO-III. We detected CD133 expression by using quantitative real-time polymerase chain reaction and Western blot and found that expression of CD133 was upregulated in cisplatin resistance of KATO-III cells (Cis-KATO-III) compared with KATO-III cells, indicating the role of CD133 in regulating cisplatin resistance of KATO-III cells. Then we sorted the Cis-KATO-III cells into CD133-positive (CD133+) pools and measured the proliferation and apoptosis after the cell is transfected with pc-CD133 and sh-CD133 by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide assay and flow cytometry. The results showed that the inhibition of CD133 inhibited the cell viability and promoted the cell apoptosis after cisplatin treatment. Furthermore, we found that inhibition of CD133 downregulated the expression of PI3K/AKT and promoted the expression of mammalian target of rapamycin, thus inhibited the autophagic activity in the Cis-KATO-III cells after cisplatin treatment. Besides, we also verified the effects of CD133 in vivo. The results indicated that inhibition of CD133 enhanced the Cis-KATO-III cell sensitivity to cisplatin by regulating PI3K/AKT/mTOR signaling pathway. In summary, our data provide new insight that CD133 activates the PI3K/AKT/mTOR signaling transduction pathway, resulting in activation of autophagy and cisplatin resistance of Cis-KATO-III cells. These results may offer a novel therapeutic target in cisplatin-resistant gastric cancer.
Introduction
Despite the morbidity and mortality of gastric cancer (GC) has declined around the world recently, it is still the fifth common cancer and the second cause of cancer death. 1 Gastric cancer treatment is a major human challenge, and China is among the nations with the highest incidence of GC. 2 Hence, we conducted our own experiments to look into potential ways of improving current diagnosis and therapy methods, referencing the molecular mechanisms of GC.
Cisplatin, as a chemotherapy medication, was the most commonly used chemotherapeutic agent for GC treatment. 2 However, drug resistance limited the clinical effect of cisplatin. Previous study had proved that cisplatin often encountered poor therapeutic efficiency against cisplatin-resistant cancer cells, insufficient accumulation at tumors, decreased cellular uptake, increased drug efflux, DNA repair, and metabolic modification and detoxification, resulting from the disorder of membrane protein transporters and cytoplasmic enzymes. 3 Nevertheless, the molecular mechanisms of drug resistance appeared to be multifactorial and unclear. 4 Therefore, it is necessary for totally understanding the molecular mechanism involved in cisplatin resistance for better treatment of GC.
Cancer stem cell (CSC) theory suggests that tumors maintain hierarchical cell organization of normal tissues. 5 Subpopulations with stem cell–like properties sustain tumor development, growth, and dissemination via unlimited self-renewing. 6 On the contrary, although undergoing rapid division, differentiated non-stem tumor cells own a reduced self-renewal activity which may have an impact on their life span. 7 High expression of extrusion pumps and DNA repair mechanisms created the conditions for CSCs to obtain resistance to chemotherapeutic and radiation therapies. Thus, conventional antineoplastic agents often fail to eradicate CSCs and surviving stem-like cells grow in tumor mass, causing recurrence. 8 In addition, a recent paper indicated that traditional chemotherapy and radiotherapy may select and enlarge drug-resistant CSC subpopulations within tumors, promoting their malignancy. 9
Previous study has demonstrated that a series of CSC markers, such as CD133, CD44, CD24, ALDH, CDCP1, EpCAM, LGR5, and so on, play pivotal roles in tumorigenesis. 10 Lee et al showed that CD44 promoted self-renewal and circulating capacities of hepatocarcinoma cells. 11 Sagiv et al proved CD24 as a new oncogene in colorectal cancer carcinogenesis. 12 CD133 (prominin-1), a 5-transmembrane domain glycoprotein, was first identified as a marker of the hematopoietic stem cell. 13 In recent researches, CD133 played a crucial part in various types of cancers. For example, CD133 was proved to promote migration in gallbladder carcinoma. 14 Upregulation of CD133 contributed to the promotion of hepatocellular carcinoma induced by STAT3. 15 CD133+ liver tumor-initiating cells promoted tumor angiogenesis, growth, and self-renewal via NTS/IL-8/CXCL1 signaling pathway. 16 However, existing researches are inadequate to prove the underlying molecular mechanism, which needs further study to be performed on CD133 mechanism in tumor cells.
AKT serine–threonine kinase 1 (AKT) serves as a target and effector of phosphatidylinositol 3-kinase (PI3K) downstream. 17 The PI3K/AKT signaling pathway is recognized to regulate the cell growth and fate decisions in tumors. 18 For example, PI3K/AKT signaling pathway was activated in the progression of glioma. 19 Wei et al identified that PI3K/AKT signaling pathway was activated by CD133/p85 interaction and promoted tumorigenesis of glioma stem cells. 19 Song et al found CD133 activated the PI3K/AKT signal transduction pathway through direct interaction with PI3K-p85 in GC cells. 20 However, there is no research on the molecule mechanism of PI3K/AKT signaling pathway between cisplatin resistance and CD133+ GC cells properties. Autophagy is a constitutive catabolic pathway that mediates both nonspecific and targeted sequestration of cellular organelles and other macromolecules, which permits the degradation of cellular components in lysosomes and the recycling of bioenergetic metabolites. 21 -23 Mammalian target of rapamycin (mTOR), a serine/threonine kinase protein of 289 kDa, plays an important role in cellular signal transduction mediated by PI3K. 24 The activation of mTOR results in the inhibition of cell autophagy 25 and drug resistance. 26 To test the cell autophagic activity, Mizushima et al suggest to detect LC3 conversion (LC3-I to LC3-II) by immunoblot analysis. 27 Thus, LC3 immunoblot analysis was applied to detect the autophagic activity in this study.
In our study, the expression of CD133 in cisplatin-resistant GC cells was evaluated. Moreover, the regulation of CD133 on cisplatin resistance via cell proliferation, apoptosis, and autophagy was elucidated. Meanwhile, we analyzed key proteins in the PI3K/AKT/mTOR signaling pathway to expand the molecular mechanism for cisplatin resistance induced by CD133 in GC cells.
Materials and Methods
Cell Culture and Reagents
The human GC cell line KATO-III was purchased from BeNa Culture Collection (Beijing, China). KATO-III cells were incubated in RPMI-1640 (Thermo Fisher Scientific, Waltham, Massachusetts) containing 10% fetal bovine serum. Cisplatin-resistant KATO-III cells (Cis-KATO-III) were obtained from KeyGEN BioTECH (Nanjing, Jiangsu, China). Briefly, cisplatin was added at increasing concentrations, with the initial concentration being 1 μg/mL, and every 4 weeks, the cisplatin concentration was increased by 1 μg/mL. The final concentration of cisplatin was 5 μg/mL. Following the experimental instructions, these Cis-KATO-III cells were incubated in culture medium containing 500 ng/mL cisplatin (Thermo Fisher Scientific) to establish normal cell growth. All cell lines were cultured in a moist atmosphere at 37°C with 5% CO2. For cisplatin and rapamycin treatment, 10 μM cisplatin and 5 μM rapamycin were extra added to the medium, respectively.
Cell Apoptosis Analysis
All the apoptotic cells were detected via an Annexin V-FITC/Propidium iodide (PI) Apoptosis Detection Kit of Thermo Fisher Scientific. Briefly, after cisplatin treatment for 24 hours, 5 × 105 cells were harvested to incubate with Annexin V-FITC and PI for 15 minutes and then prepared for subsequent analysis. Apoptosis rate was detected in comparisons with nontreated control cells. Applied Beckman Coulter’s Epics XL (Shanghai, China) instrument, apoptosis cells at early and advanced stages were counted as the total apoptotic cells, which were positive to Annexin V-FITC alone, both Annexin V-FITC and PI, respectively. The experiments were performed 3 times following the manufacturer’s instruction.
Flow Cytometry and Cell Sorting
Gastric cells were tested for a panel of fluorescent-labeled monoclonal antibodies and respective isotype controls after incubation for 48 hours. After being washed, the labeled cells were analyzed by flow cytometry with a FACS Vantage cell sorter (Becton & Dickinson, Mountain View, California). The antibodies used were CD133 (rabbit, ab216323, 1:2000; Abcam, Cambridge, Massachusetts). Sorted cells were resuspended by 1× phosphate-buffered saline (PBS).
Cell Viability
After 1 × 104 GC cells were plated on each well of 96-well plates for 24 hours, they were immersed in PBS or different cisplatin concentrations for 72 hours as control group and treatment group, respectively. Following the incubation with the Cell Counting Kit-8 (CCK-8) solution (Dojindo, Kumamoto, Japan) from the volume ratio 1 to 10 at 37°C for 90 minutes, the absorbance at 450 nm was calculated by Tecan Infinite M200 PRO (Grödig, Austria). The vacant background absorbance due to the cell absence was subtracted. The absorbance proportion of cisplatin-treated cells was defined as cell viability. The half-maximal values of inhibitory concentration (IC50) value of cells was tested by nonlinear regression analysis with the utilization of SPSS 17.0.
RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
All the RNAs were extracted by TRIzol reagent (Invitrogen, Carlsbad, California) based on the instructions. Before converting to complementary DNA, the concentration and quality of these isolated RNA samples were measured by NanoDrop 2000 (Thermo Fisher Scientific). SYBR Green I Master (Roche, Eugene, Oregon) in a LightCycler 480 (Roche) was employed to conduct quantitative real-time polymerase chain reaction (qRT-PCR) with the primer pairs in Table 1. The fold changes related to the controls were calculated by 2−△△CT method.
Sequence Information of PCR Primers.

Western Blot
Collected and lysed in Radio-immunoprecipitation assay (RIPA) buffer (Beyotime, Haimen, China), cells were mixed with a phosphatase inhibitor (Roche) and a protease inhibitor phenylmethanesulfonyl fluoride (Beyotime). The protein was analyzed by bicinchoninic acid protein assay kit (Beyotime). Total protein was divided half-and-half by sodium dodecyl sulphate–polyacrylamide gel electrophoresis and moved onto polyvinylidene difluoride membranes (Millipore, Bedford, Massachusetts), which were then blocked in 5% dry non-fat milk at room temperature for 60 minutes and incubated with the desired primary antibodies overnight at 4°C, which were CD133 (rabbit, ab216323, 1:2000; Abcam), LC3 (rabbit, ab48394, 1:1000), p-PI3K (rabbit, ab182651, 1:1000; Abcam), PI3K (rabbit, ab40755, 1:1000; Abcam), p-AKT (rabbit, ab81283, 1:5000; Abcam), AKT (rabbit, ab8805, 1:500; Abcam), Bcl-2 (rabbit, ab32124, 1:500; Abcam), Bax (rabbit, ab32503, 1:1000; Abcam), mammalian target of rapamycin (mTOR) (rabbit, ab2732, 1:2000; Abcam), phospho-mTOR (p-mTOR) (rabbit, ab109268, 1:1000; Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (rabbit, ab8245, 1:500; Abcam). After washing, the secondary antibodies (goat anti rabbit, ab97080, 1:5000; Abcam) were added at room temperature and cultured for 1 hour. Finally, all the protein bands were evaluated by enhanced chemiluminescence detection kit (Millipore). GAPDH acted as a loading control.
Transfection
To be homologous to CD133, three different sequences of shRNA were designed by lentiviral expression vector (pGreenPuro Vector; SBI [Mountain View, California]). Target sites of encoding CD133 as well as a negative control sequence in human genes are listed in Table 2. We transformed the Escherichia coli strain DH5α using each DNA that was purified with a Qiagen End-Free Plasmid Mega Kit (Qiagen, Valencia, California). Polymerase chain reaction amplification and sequencing were applied to verify the ligation product.
Sequence Information of shRNA.

Immunofluorescence
Immunofluorescence staining assay was conducted to observe the expression level of microtubule-associated protein 1 light chain 3 (LC3) in Cis-KATO-III. Twenty-four hours after transfection, the samples were washed by PBS and fixed by 4% paraformaldehyde overnight at 4°C, then these fixed samples were rinsed again with PBS and permeabilized with 0.2% Triton X-100 in PBS at 37°C for 10 minutes. After being blocked with 1% bovine serum albumin at 37°C for 1 hour, samples were treated with rabbit anti-LC3 antibody (ab51520, 1:3000; Abcam) overnight at 4°C. Then samples were washed by PBS for 3 times and incubated in goat anti-rabbit IgG (H + L) (ab150077, 1:500; Abcam) for 1 hour. After rinsed with PBS, they were further stained with DAPI (4′,6-diamidino-2-phenylindole) for 1 hour and then imaged using a confocal microscope.
Immunohistochemical Assays
Fresh tissue samples were deparaffinized by xylene and dehydrated through a graded alcohol series. Endogenous peroxidase activity was interrupted in methanol with 0.5% H2O2 for 10 minutes. For antigen retrieval, sections were microwaved in 0.01 M sodium citrate (pH 6.0). The sections were cultured in PBS with 10% normal goat serum for 1 hour at room temperature to prohibit the nonspecific binding. Without washing, these sections were cultured with the Ki67 antibody (rabbit, ab15580, 1:200; Abcam), which has been validated by previous studies, in PBS at 4°C overnight in a moist box and then incubated with secondary antibody biotinylated goat anti-rabbit (ab6746, 1:500; Abcam) for 1 hour at room temperature. The Ki67 expression was detected by streptavidin–peroxidase complex. The brown color indicative of peroxidase activity was developed through incubating the sections with 0.1% 3, 3-diaminobenzidine in PBS containing 0.05% H2O2 for 5 minutes at room temperature.
Animal Model
It was separated into 5 groups and 5 mice in each group randomly for 25 male BALB/c nude mice of 5 weeks old without thymic (Laboratory Animal Center of Shanghai, Shanghai, China). The nude mice were injected approximately 5 × 106 transfected CD133+ Cis-KATO-III cells on the right flanks. Cisplatin and rapamycin were given by injecting 5 mg/kg intraperitoneally with a single dose of 10 and 5 μM, respectively, once a day, for 7 consecutive days. The mice were killed at 4 weeks after inoculation. The tumor volume was measured every 7 days by the longest and shortest tumor diameters with calipers and calculated as: (shortest diameter)2/2 × longest diameter. Expression of LC3-II/I/PI3K/AKT/mTOR/Bax/Bcl-2 was analyzed by Western blot method. Expression of Ki67 was analyzed by immunohistochemical (IHC). Animal studies and procedures were based on institutional guidelines.
Statistical Analysis
SPSS software 17.0 (IBM, New York, New York), GraphPad Prism software 6.0 (GraphPad, San Diego, California, USA), Student t test, and one-way analysis of variance after Tukey test were applied to all the statistical analysis. Data were displayed as the means ± standard deviation. P < .05 were considered as statistically significant.
Results
Enrichment of CD133+ Cells in Cis-KATO-III
Cisplatin sensitivity was assessed via CCK-8 assay following 48 hours of treatment, and this assessment showed that Cis-KATO-III cells were more resistant to cisplatin than parental KATO-III cells (Figure 1A, **P < .01). The cisplatin IC50 were exhibited (Figure 1B, **P < .01). The different expressions of CD133 among these cells were supplied by qRT-PCR and Western blot. In contrast to KATO-III cells, cis-KATO-III cells displayed higher levels of CD133 messenger RNA and protein expression (Figure 1C-D, **P < .01). We further evaluated the protein expression of PI3K/AKT/mTOR in the two cells. The results showed the expression of PI3K and AKT was higher in Cis-KATO-III cells than in the KATO-III cells, while the expression of mTOR was lower in Cis-KATO-III cells than in the KATO-III cells (Figure 1E, **P< 0.01). Flow cytometry analysis demonstrated increased proportions of CD133+ cells in cis-KATO-III cells (Figure 1F, **P < .01). CD133+ KATO-III cells were identified to be CD133 positive (Figure 1G, P > .05) and be further used for following experiments.

CD133 overexpressed in gastric cancer and especially in cisplatin-resistance gastric cancer stem-like cells. A, Effects of cisplatin on viability of KATO-III and Cis-KATO-III cells. Cells were treated with gradient concentrations of cisplatin for 48 hours, respectively, and subsequently performing Cell Counting Kit-8 (CCK-8) assay to quantified cell viability. B, Half-maximal values of inhibitory concentration (IC50) values in response to cisplatin treatment were examined via CCK-8 assay. C, Relative CD133 mRNA expression in KATO-III and cis-KATO-III cells was analyzed by quantitative real-time polymerase chain reaction. D, Western blot results showed a relative expression of CD133 protein in KATO-III and cis-KATO-III cells. E, Western blot results showed relative expression of PI3K, AKT, mTOR protein in KATO-III and cis-KATO-III cells. F, Flow cytometry separated CD133− as well as CD133+ cells and showed the percentage of CD133+ cells in KATO-III and Cis-KATO-III cells respectively. G, Flow cytometry results showed the percentage of CD133+ cells in KATO-III and Cis-KATO-III cells after sorted, respectively. Data are presented as the mean ± standard deviation of triplicate experiments. *P < .05, **P < .01.
Function of CD133 in Cis-KATO-III
We transfected shRNA of CD133 and successfully knocked down the expression of CD133, while pc-CD133 successfully overexpressed CD133 in CD133+ Cis-KATO-III cells (Figure 2A and B, **P < .01). Knock down of CD133 significantly increased apoptosis and inhibited cell proliferation while transfection with pc-CD133 received opposite results (Figure 2C and D, *P < .05, **P < .01). Expression of PI3K/AKT signaling pathway-related protein and apoptosis-related protein was analyzed by Western blot and indicated that CD133 activated PI3K/AKT signaling pathway while inhibited apoptosis (Figure 2E, **P < .01).

Function of CD133 in cis-KATO-III cells. Three CD133− targeted short hairpin RNA (shRNA) as well as pc-CD133 were established and transfected into CD133+ cisplatin-resistant KATO-III and the expression of CD133 was analyzed by quantitative real-time polymerase chain reaction (A) and Western blot (B), and GAPDH was used as an internal control. C, Apoptosis ratio of 3 groups of cells was analyzed by flow cytometry. D, Cell Counting Kit-8 analysis results of cell proliferation. E, Activation of PI3K/AKT signaling pathway and expression of apoptosis-related protein Bcl-2 and Bax was analyzed by Western blot, and GAPDH was used as an internal control. Data are presented as the mean ± standard deviation of triplicate experiments. *P < .05, **P < .01.
Downregulation of CD133 Reversed Cisplatin Resistance
To validate the relation between CD133 and cisplatin resistance, CD133 overexpression and knockdown were utilized. The IC50 values of transfection of sh-CD133 cells were decreased and increased in pc-CD133 group compared with NC (Figure 3A and B, **P < .01). Ten micrometers of cisplatin was incubated with CD133+ Cis-KATO-III for further experiments. Cell viability assay showed that the effect of cisplatin on cell proliferation was improved after knocking down CD133 (Figure 3C, *P < .05, **P < .01). The percentage of apoptotic cells in CD133 knocked down cells exposed to cisplatin also increased compared with the NC group (Figure 3D, **P < .01). To figure out the molecular mechanism on how CD133 increased KATO-III properties and promoting cisplatin resistance, Western blot confirmed the activation of PI3K/AKT and the regulation of Bax and Bcl-2. To be specific, sh-CD133 improved the effect of cisplatin on inhibition of Bcl-2 and PI3K/AKT signaling pathway as well as the promotion of the expression of Bax (Figure 3E, **P < .01).

Downregulation of CD133 reversed the effect of cisplatin in cisplatin-resistant cells. A, Effects of cisplatin on the viability of Cis-KATO-III cells transfected with sh-CD133 or pc-CD133. Cells were treated with gradient concentrations of cisplatin for 72 hours, respectively, and subsequently performing Cell Counting Kit-8 assay to quantify cell viability. B, Half-maximal values of inhibitory concentration (IC50) values in response to cisplatin treatment were examined via CCK-8 assay. C, Cell Counting Kit-8 analysis of cell viability. D, Apoptosis ratio of 4 groups of cells was analyzed by flow cytometry. E, Activation of PI3K/AKT signaling pathway and expression of apoptosis-related protein Bcl-2 and Bax was analyzed by Western blot, and GAPDH was used as an internal control. Data are presented as the mean ± standard deviation of triplicate experiments. *P < .05, **P < .01.
CD133 Increased Autophagy via Inactivating mTOR Signaling Pathway
For further molecular mechanism between CD133 and cisplatin resistance in Cis-KATO-III, autophagy-related protein LC3II/I and mTOR expression was measured by Western blot (Figure 4A, *P < .05, **P < .01). Fluorescence intensity of GFP-LC3 was also shown (Figure 4B). These illustrated that downregulation of CD133 increased the suppression of cisplatin in Cis-KATO-III. We also treated Cis-KATO-III cell lines with rapamycin and the results indicated that cisplatin treatment significantly inhibited autophagy in CD133 knocked down cell line, compared with the NC group and rapamycin group, respectively.

Autophagy was increased by CD133 via mTOR signaling pathway. A, Expression of mTOR and LC3II/I was analyzed by Western blot, and GAPDH was used as an internal control. B, Fluorescence intensity of GFP-LC3 was shown. Rapamycin was control groups for autophagy. Data are presented as the mean ± standard deviation of triplicate experiments. *P < .05, **P < .01.
CD133 Promoted Tumorigenesis and Autophagy In Vivo
Furthermore, we subcutaneously injected 4 groups of CD133+ Cis-KATO-III to nude mice to explore how CD133 affect cisplatin resistance in vivo. Tumor photograph, volume, and weight are shown (Figure 5A-C, *P < .05, **P < .01). Results showed that downregulation of CD133 or orally given cisplatin led to smaller and lighter tumor, while co-treatment with sh-CD133 and cisplatin significantly inhibited tumor formation. We also used IHC to detect that Ki67 expression was downregulated by co-treatment of sh-CD133 and cisplatin, indicating that reduction in CD133 significantly reversed cisplatin resistance in Cis-KATO-III (Figure 5D). In addition, detection in the expression LC3II/I indicated that downregulation of CD133 or orally given cisplatin resulted in the reduced autophagy activity in vivo, and co-treatment of sh-CD133 and cisplatin significantly inhibited autophagy activity in vivo (Figure 5E, **P < .01). Also, Western blot results showed that inhibition of CD133 reduced the expression of PI3K/AKT/Bcl-2 and promoted the expression of mTOR/Bax in vivo (Figure 5E, **P < .01). These results indicated that inhibition of CD133 overcomes cisplatin resistance through inhibiting PI3K/AKT/mTOR signaling pathway in vivo.
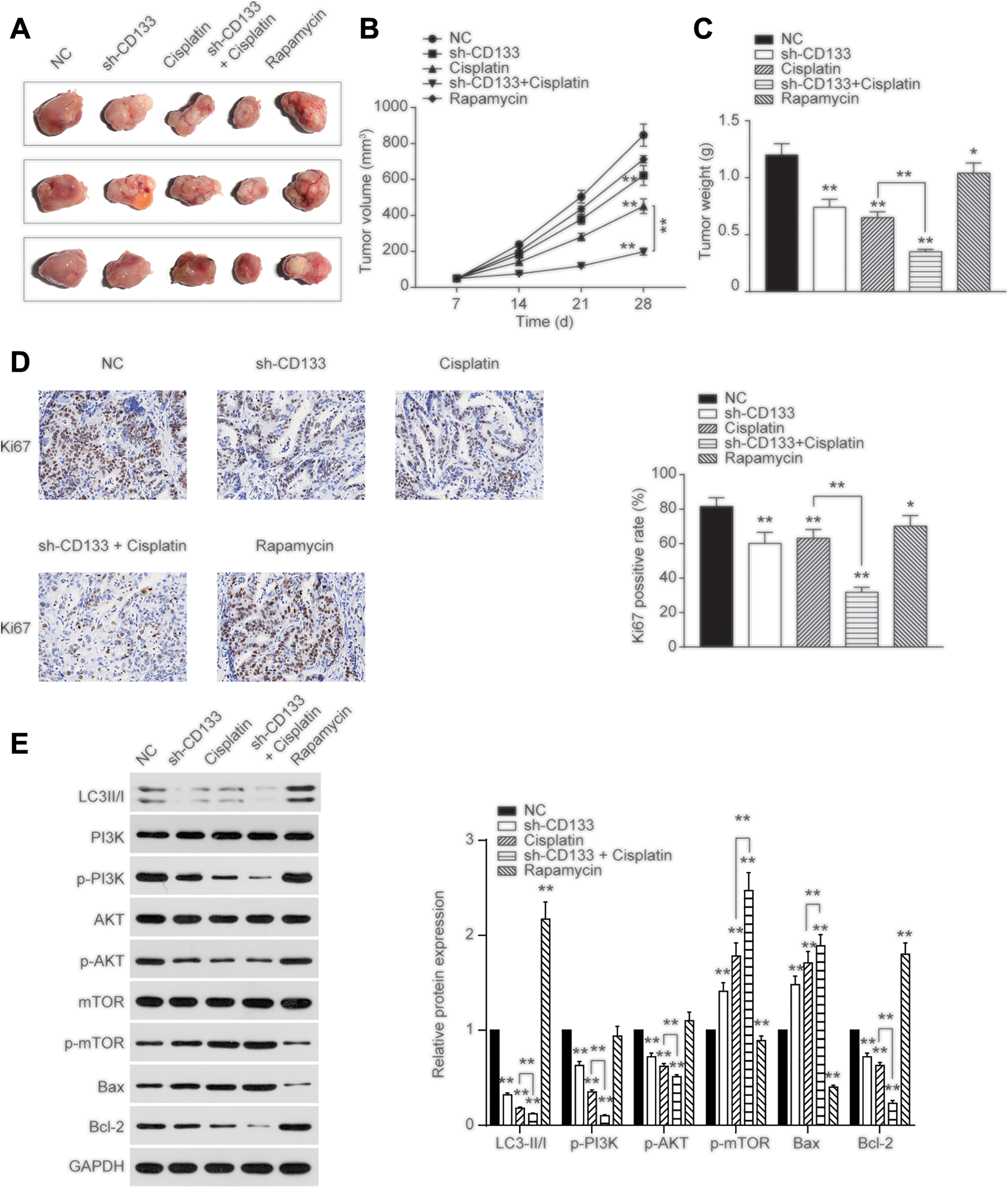
CD133 promoted tumorigenesis in vivo. A, Tumor was established by injecting CD133+ cis-KATO-III cells subcutaneously and respectively. Representative photographs of tumors were shown. B, Tumor volume was assessed every 7 days and shown. C, Tumor weight was analyzed at day 28. D, Immunohistochemical (IHC) results showed Ki67 expression in tumor tissues. E, Expression of LC3II/I/PI3K/AKT/mTOR/Bax/Bcl-2 was determined by Western blot, and GAPDH was used as an internal control. Data are presented as the mean ± standard deviation of triplicate experiments. *P < .05, **P < .01.
Discussion
In our study, we established cisplatin-resistant GC cells and separated CD133− or CD133+ cells. Results on CD133+ cisplatin-resistant KATO-III cell line showed that CD133 promoted cell proliferation, antiapoptosis, and autophagy and finally inhibited the therapeutic efficiency of cisplatin. Meanwhile, activation of PI3K/AKT/mTOR signaling pathway and expression of apoptosis-related protein were verified to contribute to the promotion of CD133 on cisplatin resistance.
Much evidence revealed that CSCs in tumors might have the responsibility for the introduction and progression of neoplastic tissue. 5 Cancer stem cells were more resistant to chemotherapy due to their capacity for activating DNA repair and evading apoptosis, which might be the reason of cancer relapse after traditional therapies. 8 CD133, a marker on the cell surface, first identified the CSCs in cerebral tumors. 28 After that, increasing studies demonstrated that there was CSC property in CD133+ cell subpopulations isolated from tumors. For instance, CD133 accelerated tumor progression in hepatocellular carcinoma as a crucial factor of CSCs, 29 ovarian cancer, 30 and glioma cancer. 31 CD133+ was identified as the marker of chemoresistance in breast cancer. 32 Bertolini et al confirmed that CD133+ cells were enriched in lung cancer by in vitro cisplatin treatment and in clinic patient samples. 33 Consistently, our study discovered that CD133 expression and percentages of CD133+ cells were increased in cisplatin-resistant cells compared with normal GC cells. CD133+ promoted cell proliferation as well as inhibited apoptosis, which promoted CSC properties. Animal model induced by subcutaneously injected CD133+ Cis-KATO-III evidenced the function of CD133.
Cisplatin resistance reduced the therapeutic efficiency in many cancers, while the molecular mechanism of cisplatin resistance was complex and unclear. Several studies had investigated the relationship between CD133 and cisplatin resistance in CSCs. It was reported that CD133+ non-small cell lung cancer cells showed significant resistance to cisplatin compared with CD133− cells. 34 Zhang et al confirmed that inhibited CD133+ CSCs enhanced the effectiveness of cisplatin in laryngeal carcinoma treatment. 35 Similarly, we transfected sh-CD133 in CD133+ cisplatin-resistant GC cells and results showed that downregulation of CD133 promoted sensitivity to cisplatin. The results indicated that CD133 played a vital role in GC cisplatin resistance.
Yu et al confirmed that PI3K/AKT/mTOR signaling pathway implicated in diverse cell processes of human small-cell lung carcinoma. 36 Several studies documented that CD133 could inhibit apoptosis and induce chemotherapy resistance by activating PI3K/AKT signaling pathway in GC and hepatocellular cancer cells. 37 Autophagy was demonstrated to play a key role in CD133 regulating drug resistance, 38 and activated mTOR signaling pathway decreased autophagy and improved drug therapeutic efficiency. 39 Similarly, we pointed out that PI3K/AKT/mTOR signaling pathway was remarkably inhibited by co-incubation with sh-CD133 and cisplatin in CD133+ cisplatin-resistant GC cells compared with NC group as well as single transfection of sh-CD133 or treatment with cisplatin, implying that PI3K/AKT/mTOR signaling pathway was important in CD133-mediated cisplatin resistance. Therefore, we indicated that CD133 might improve GC cisplatin resistance in the tumor microenvironment by upregulating PI3K/AKT/mTOR signaling pathway.
Nonetheless, results of xenografts reveal that the expression changes of Bcl-2 and Bax between cisplatin group and cisplatin/sh-CD133 group in vivo were lower than that in in vitro results. As pro-apoptotic proteins, the activities of Bcl-2 and Bax are precisely regulated by a complex network of proteins, simultaneously healthy cells can survive by constant control of dynamic translocation of these proteins between mitochondria and cytosol. 40 In this research, xenograft assays were performed in mice with tumor formation, which still have healthy cells that have the capability to control the shuttling of pro-apoptotic proteins. In addition, in view of the remarkable tissue otherness between human and mice, these above results in vivo and in vitro both have considerable reference value.
There are still some limitations in our study. We chose only one GC cell line and corresponding cisplatin-resistant cell line. Detailed mechanisms of CD133 regulating resistance symptoms needed further research. In Cis-KATO-III cells, the factors which affect the CD133 expression should be further investigated.
In conclusion, CD133 was found to promote gastirc cancer stem cells' (GCSCs) cisplatin resistance by increasing cell proliferation, anti-apoptosis, and autophagy abilities via activating PI3K/AKT/mTOR signaling pathway. Knocking down of CD133 might act as a potential mean to reverse cisplatin resistance in the therapy of cisplatin-resistant GC.
Footnotes
Authors’ Note
Our study was approved by the ethical committee of Shanghai University of Traditional Chinese Medicine, East Hospital of Tongji University (Approval No. PZSHUTCM190315023). All animal housing and experiments were conducted in strict accordance with the institutional Guidelines for Shanghai University of Traditional Chinese Medicine, East Hospital of Tongji University.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China (91570577).