
Abstract
In this study, hydrophilic based bioactive nanofibers were produced via an electrospinning and electrospraying simultaneous process. Poly(vinyl alcohol) (PVA), poly(vinyl alcohol)-gelatin (PVA-Gel), and poly(vinyl alcohol)-alginate (PVA-Alg) polymers were used as the matrix material and folic acid (FA) particles were dispersed simultaneously on the surface of the nanofibers. The morphology of the nanofibers (NFs) was uniform and confirmed by scanning electron microscopy. Thermal behavior, chemical structure of the composite nanofibers were investigated by thermogravimetric analysis, and Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy which showed that no chemical bonding between vitamin and polymers. A fast release of FA-loaded electrospun fibers was carried out by UV-Vis in vitro study within the 8 hour-period in artificial sweat solutions (pH 5.44). The obtained PVA/FA, PVA-Gel/FA, and PVA-Alg/FA fibers released 49.6%, 69.55%, and 50.88% of the sprayed FA in 8 h, indicating the influence of polymer matrix and polymer-drug interactions, on its release from the polymer matrix. Moreover, biocompatibility of all developed novel NFs was assessed by two different cytotoxicity tests, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and neutral red uptake (NRU) assay in L929 (mouse fibroblasts) cell lines. In all cases, it is concluded that these new electrospun fibers had fast-release of the vitamin and the hybrid process is suitable for transdermal patch applications, especially for skin-care products. The results of cytocompatibility assays on L929 reveal that all prepared NFs have no or slight cell toxicity. PVA and PVA-Gel with/without FA nanofibers seems more biocompatible than PVA-Alg nanofibers.
Introduction
Human skin, the largest organ in the body, has vital functional role by protecting from pollution, ultraviolet radiation and other damage, which cause to skin aging and disorders [1–3]. Skin has well-organized morphological structure and self-renewing barrier property. It comprises of three multilayers structure with different levels of cellular and epidermal differentiations that serves as a protective barrier [4,5]. The outmost layer of the skin is epidermis known as the stratum corneum (SC) contains dead cells (corneocytes) interdispersed within a greasy matrix [1,2,6]. Therefore, SC provides selective permeability and limits hydrophilic rich compounds to penetrate into the inner layers.
Transdermal drug delivery system (TDDS) is defined as pharmaceutical ingredients are transported locally or systemically via skin [1,7,8]. The approach is alternative to oral, intravascular, subcutaneous, and transmucosal routes due to loss of effectiveness and toxicity of drugs (degradation) [9,10]. Besides, TDDS has not cause painful administration and other unwanted side effects on the gastrointestinal tract compared with oral or injection administration, especially. Therefore, this route has not damage to liver. However, the penetration of drugs via SC has many limitations. It is known that drug molecules penetrate pass through SC by two routes including intercellular (hydrophilic drugs) and intracellular (hydrophobic drugs) mechanisms. A drug molecule needs some physicochemical properties such as sufficient hydrophobicity, high and sufficient partition coefficient (LogP 1–3), low molecular weight (<500 Da) and the short half life for better penetration [1,11,12].
Folic acid is an important member of B-complex vitamins, which is known as B9 vitamin [13,14]. It consists of 4-[(pteridine-6-ylmethyl)-amino] benzoic acid skeleton conjugated with one or more L-glutamate units [15,16]. Folic acid plays a crucial role in maintenance many metabolic functional processes in the body, such as cell growth, and development, synthesis of nucleic acids and metabolism of many amino acids [14,17]. Many topical formulations containing folic acid are available on the market for the treatment of damaged or aged skin. But topical formulations such as cream and gels containing folic acid tend to accumulate in the skin with a lipophilic structure. Moreover, it is often ignored that folic acid is more sensitive to sun light than retinol, thiamine and niacin vitamins [18]. As a results of prolonged exposure of folic acid to sun light, two reactive products consisting of 2-amino-4-hydroxy-6formyl pteridine (pterin-6-carboxylate) and paminobenzoyl-L-glutamic acid are released. Then, pterin-6-carboxyaldehyde is converted to pterin-6-carboxylic acid (PCA) and then to decarboxylated 2-amino-4-hydroxy pteride. Pterin–6-carboxyaldehyde and PCA can cause the production of reactive oxygen species responsible for DNA damage and cytotoxicity [19]. For this reason, the use of folic acid by encapsulating in pharmaceutical, cosmetic and biological applications can facilitate its use.
PVA, gelatin and alginate are biocompatible, hydrophilic, and water soluble polymers [20,21]. The blends of PVA with gelatin and alginate have been also reported for using drug delivery systems [22–24].
During the last decades, nanofibers have become an excellent candidate for transdermal drug release applications due to high porosity, large specific surface area and small size of electrospun fibers [8,25]. There have been many attempts about pharmaceutical ingredients incorporated into nanofibers for usage of transdermal applications. In this regard, Manatunga et al. reported polyethylene oxide and gelatin (PEO-Gel) fibers with Au nanoparticles can be potential beauty mask [26]. Gencturk et al. fabricated polyurethane/hydroxypropyl cellulose electrospun nanofibers with Donepezil (DNP) hydrochloride and in their study, controlled release of DNP from the fibers within 6 h was observed [27]. With the similar strategy, Esentürk et al. have investigated in vitro drug release behavior of voriconazole (VCZ) loaded hydrophilic poly(vinyl alcohol)/sodium alginate (PVA/SA) electrospun NFs [28].The obtained NFs indicated sustain release behavior with 8 h. Also in a similar study reported by Nematpour et al. that tetracycline (TC) loaded polycaprolactone (PCL) and graphene fibers were prepared by electrospinning, the resulting materials released the drug with sustained behavior during 120 h [29].
Abdoli et al. developed transdermal patch based on PVA/gum tragacanth/graphene oxide electrospun composite fiber for skin treatment [30]. These NFs showed cumulative drug release reached to 95% after 48 h.
Kataria et al. developed transdermal patch based on ciprofloxacin-loaded PVA and sodium alginate (NaAlg) electrospun composite nanofiber for wound healing [22]. In another study, Tran et al. investigated release behavior of Ibuprofen from NFs. Polycaprolactone (PCL), poly(N-isopropylacrylamide) pNIPAM and pNIPAM/PCL were used as carriers of the drug for thermo-responsive transdermal delivery systems [31]. The release behavior of the fibers significantly changed with 4-hour period depending on the temperature.
Furthermore, vitamin loaded NFs have received increasing attention as transdermal patches. Within this aim, Madhaiyan et al. synthesized vitamin B12-loaded polycaprolactone NFs and the resulting fibers indicated gradually vitamin release behavior in 48-hour period [32]. A controlled release of Zingiber cassumunar Roxb oil was observed from PVP NFs over 50 hours to utilize for skin care applications [33].
Barani et al. produced PVA nanofibers containing thiosemicarbazone in core-shell forms [34]. The drug molecules were preserved by shell PVA matrix. However, the core-shell fibers showed low drug release rate as low as 17%. Immobilize of the bioactive substances into fabrics can also be used with a similar perspective in the fabrication of medical textiles [35]. Parin et al. fabricated honey-loaded PVA nanofibers to use biomedical purposes [20].
Previous studies mainly focused on bioactive material-loaded NFs fabricated by blending and coaxial (core-shell) electrospinning. Apart from electrospinning techniques, there is one approach used in drug release applications which is called electrospraying. Electrospraying is sister technology of electrospinning to fabricate nanomaterials with <1 μm diameter [36]. In electrospinning, concentration of polymer solution is sufficient to develop chain formation in the capillary by using high voltage potential while the polymer solution concentration is too low and droplets is spraying from the capillary in electrospraying [37]. However, nanoparticles micro/nanoparticle dispersions are processed with electrospraying due to low viscosity. Generally, the bioactive substances could be dispersed directly in polymer solution to obtain nanofibers, but there is a limitation of particle aggregation in the polymer solution in this process [38]. Unlike other electrospinning methods, such as blending, emulsion and core-shell, drug molecules can both penetrate into fibers and hold on surface of fibers, as well. There are many studies on nanofibers produced together with electrospinning and electrospraying process. However, to the best of our knowledge, there is no report on the transdermal drug delivery of nanofibers produced with simultaneous electrospinning and electrospraying process.
In the current study, electrospinning and electrospraying methods were performed simultaneously to fabricate the electrospun PVA, PVA-Gel and PVA-Alg fibrous mats. Folic acid was utilized as a model drug. Folic acid was encapsulated between and inside the nanofibers to produce produced different fibers and also it has been aimed to investigate the availability of folic acid as a beauty mask in release applications. By varying the polymer matrix type, the properties of as-spun/FA NFs were investigated in terms of morphology, thermal and chemical structure (polymer-polymer and polymer-drug compatibility). In vitro drug release from the NFs was assessed. The cytocompatibility of the nanofibers was also evaluated through3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and neutral red uptake (NRU) assays on the L929 cell lines (mouse fibroblasts) as recommended in ISO10993-5 standard.
Materials and methods
Materials
The Poly(vinyl alcohol) (PVA) (density 0.4–0.6 g/cm3, purity 87.8% and Mw ∼30.000 g/mol) was purchased from ZAG Industrial Chemicals (İstanbul, Turkey). Gelatin from bovine skin (225 Bloom, type B, BioReagent), sodium alginate (medium molecular weight) and sodium bicarbonate (NaHCO3) (99.7% purity) and acetic acid (CH3COOH) with 80% purity obtained from Sigma Aldrich Chemical Company (USA) and ethanol (99.9% purity, Tekkim). Folic acid (C19H19N7O6) for Biochemistry (98–102% purity) was supplied from ChemSolute Company (Germany). To prepare the artificial sweat solutions, L-Histidine monohydrochloride monohydrate (C6H9O2N3.HCI.H2O) (>99% purity), sodium chloride (>99.5% purity) and sodium dihydrogen phosphate dihydrate (NaH2PO4.2H2O) were purchased from Sigma Aldrich Chemical Company (USA). The chemicals used in the MTT and NRU assay tests: RPMI (Roswell Park Memorial Institute) 1640 medium, fetal bovine serum (FBS), penicillin-streptomycin, neutral red (NR) (3-amino-7-dimethyl-amino-2-methylphenazine hydrochloride), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), trypan blue, dimethyl sulfoxide (DMSO) from Merck; Triton X-100, trypsin–EDTA, Dulbecco’s phosphate buffered saline (DPBS) from Gibco. Distilled water was used in the experiments and all of the reagents were used without any purification.
Fabrication of hybrid nanofibers
The hydrophilic based polymer/FA nanofibers were fabricated by combining electrospinning and electrospraying process (INOVENSO Nanospinner24, Turkey). PVA powders were dissolved in pure water at 90°C till a 10% (w/w) homogeneous PVA solution was obtained. Gelatin was dissolved in pure water and acetic acid binary-solvent systems (7:3 w/w) solution to obtain a 20% (w/w) gelatin solution by stirring for 2 hours at ambient conditions. Alginate powders were also dissolved in water to obtain a 2% (w/w) alginate solution. PVA-Alg (4:1 v/v) and PVA-Gel solutions (4:1 v/v) were mixed at room temperature for overnight. In our previous experiments, it was determined optimum folic acid (FA) concentration in electrospraying system. 22 mg FA was dispersed in 10 mL (2:1 v/v) pure water and alcohol solution for each polymer system via ultrasonic homogenizer (Bandelin/Sonoplus HD3200). Each polymer solution (PVA, PVA-Gel and PVA-Alg) and FA particle dispersion were transferred into two separate plastic syringes (10 mL) and put side to side on two syringe pumps. The plastic syringes were attached to two stainless nozzle. The distance between two nozzles was 15.04 mm. The PVA, PVA-Gel and PVA-Alg solutions and FA dispersion were dispensed via nozzle which was vertical to therotating collector. The feed rates were performed at 2 mL/h and 1.5 mL/h for polymers and FA dispersion, respectively. Neat PVA, PVA-Gel and PVA-Alg solutions were prepared as a control (Figure 1). The process parameters used in nanofiber fabrication are shown in Table 1.

Schematic illustration of hybrid process of nanofibers.
Process parameters for electrospinning.

Nanofiber characterization
Scanning electron microscopy (SEM)
Morphology of all nanofibers was observed with Carl Zeiss/Gemini 300 Scanning Electron Microscope (SEM) (ZEISS Ltd., Germany). All samples were coated with gold for 20 minutes before analysis. Fiber diameters were measured by using Image J (version 1.520 software) by randomly selecting the diameters of 60 individual fibers for each sample. Moreover, porosity and pore size distributions were measured using SEM images of the resulting fibers by Image J.
Thermal analysis (TGA)
TA/SDT650 TGA (USA) were used for the thermal analysis. TGA analyses were performed under nitrogen atmosphere with 20°C min−1 heating rate, 30°C–600°C temperature range and applied oxygen atmosphere with 20°C min−1 heating rate 600°C–900°C temperature.
Fourier transform infrared spectroscopy (FT-IR)
FT-IR data were obtained with a Thermo Nicolet iS50 FT-IR (USA) spectrometer with a ATR (Attenuated Total Reflectance) adapter (Pike, USA) in the range of 4000–500 cm−1 recorded with 16 scans at 4 cm−1 resolutions.
In-vitro release study
The pH of skin surface ranges from 4.2 and 5.6 [39]. Furthermore, solubility of folic acid as a drug is maximum at pH 5 to 6 [40]. Therefore, artificial sweat solutions were prepared according to ISO 105-E04:2013 method [41]. The vitamin-release behavior of FA sprayed resulting fibers were studied in acidic sweat solutions at pH 5.44 by total immersion method [31]. 25 cm2 of the nanofibers were put into sealed glass tubes with each containing 50 mL of acidic sweat solution, separately. Then, they were placed in shaking incubator at 37°C with stirring 120 rpm in order to apply the release profile of the folic acid. Samples of 3 mL was removed at the specified time intervals with the sweat solution and the corresponding absorbance value was determined in a UV spectrophotometer (Scinco/NEOYSY 2000) at λmax = 282 nm, which was the characteristic peak of folic acid. The drug concentration was obtained from the calibration curve of the model vitamin prepared with a folic acid solution of known concentrations in acidic solutions (pH 5.44). The calibration curve was found to be Y = 0.0486X + (−0.0402) (R2 = 0.99992), where X is the concentration of FA (mg/L) and Y is the solution absorbance at 282 nm (linear range of 0.5–25 mg/L) (Figure S1). The amount of released drug was determined using UV–Vis spectroscopy. Three replicates for each experiment were obtained.
Entrapment efficiency (EE) and folic acid loading capacity (LC) in electrospun fibers
The folic acid entrapment efficiency (EE) was estimated as follows;
A known area (3x3 cm) of the fibers was dissolved totally in acidic sweat solution. Then, 3 mL of this solutions analyzed by a UV–Vis spectrophotometer at the characteristic wavelength of folic acid. Loading capacity of fibers (LC) were calculated to determine how much folic acid is entrapped per polymer. The EE and LC values were determined using equations (1) and (2), respectively.


In-vitro cytocompatibility
Preparation of nanofiber extract solutions for cytotoxicity assays
In this study in order to determine the cytotoxic effects of novel NFs prepared in different compositions, extracts of all samples were prepared among the methods recommended by the UNI EN ISO 10993-12: 2009 regulation and also suitable for the nature and shape of NFs as biomaterials [42]. Before performing the extraction procedure, equal sizes of NF patch samples (3 cm2) were cut and each side of all NFs were sterilized under a UV lamp (254 nm) for 1 h in a laminar flow cabinet (Telstar BioVanguard Class II Biological Safety Cabinet) in order to keep the structural properties of the nanofibers intact. The extraction procedure was carried out in sterile tubes containing 5 mL of culture medium (99% RPMI 1640 and 1% Penicillin-Streptomycin, without serum in order to prevent protein interaction). All samples were kept in this medium at 37°C for 30 minutes and it was observed that they were completely dissolved during this time period. Equal sample of an aluminum foil was also used simultaneously in the experiments as a reference base support material of NFs and treated as a sample and also extraction medium without sample was used as control and culture medium containing 1% Triton X-100 was used as a positive control.
Cell culture
All experimental parts of cell culture and cytotoxicity assays were conducted at the cell culture laboratory of Bioengineering Department of Bursa Technical University according to the good cell culture practices [43]. The L929 cell lines (mouse fibroblasts) were used for cytotoxicity assays as a reference cell line for cytotoxicity testing of medical devices and materials, according to the UNI EN ISO 10993-5rule:2009 [44]. L929 cells (kindly obtained from Yeditepe University, Department of Genetics and Bioengineering) were seeded in 75 cm2 culture flasks containing RPMI 1640 supplemented with 10% FBS and 1% penicillin streptomycin. The cells were incubated at 37°C in 5% CO2 in a humidified cell culture incubator and monitored daily by using an inverted microscope with phase contrast attachment (Olympus CKX41). Subcultures were performed when an 80% of confluence was observed. Following disaggregation with trypsin/EDTA and resuspension of cells in medium, a total of 5×104 cells/well were plated in 96 well tissue-culture plates.
Determination of the cytotoxicity of nanofibers by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT assay was performed by the method of Mosmann [45] with the modifications of Hansen et al. [46] and Kuz´ma et al. [47]. After 24 h incubation of cells seeded in 96 well tissue-culture plates, cells were exposed to the different samples of nanofiber extract solutions ((PVA) nanofiber: Poly(vinyl alcohol) (10%); (PVA-FA) nanofiber: Poly(vinyl alcohol) (10%) – Folic acid (22 mg); (PVA-Gel) nanofiber: Polyvinyl alcohol (10%) – Gelatin (20%); (PVA-Gel-FA) nanofiber: Poly(vinyl alcohol) (10%) – Gelatin (20%) – Folic acid (22 mg); (PVA-Alg) nanofiber: Polyvinyl alcohol (10%) – Alginate (2%);/(PVA-Alg-FA) nanofiber: Poly(vinyl alcohol) (10%) – Alginate (2%) – Folic acid (22 mg)) in medium and also pure folic acid (2200 µg/mL) concentration for 24 h at 37°C. After exposure, the medium was aspirated and MTT (5 mg/mL of stock in PBS) was added (10 µL/well in 100 µL of cell suspension), and cells were incubated for an additional 4 h with MTT dye. At the end of incubation period, the dye was carefully taken out and 100 µL of DMSO was added to each well. The absorbance of the solution in each well was measured in a microplate reader at 570 nm. Results were expressed as the mean percentage of cell growth from three independent experiments. Cell viability was plotted as percent of control (assuming data obtained from the absence of nanofiber as 100%).
Determination of the cytotoxicity of nanofibers by neutral red uptake (NRU) assay
The cytotoxicity of nanofibers and also pure folic acid was performed in L929 cells by NRU assay following the protocols described by Virgilio et al. [48] and Saquib et al. [49]. After 24 h incubation of L929 cells in 96 well plates, 200µL of all sample solutions were added. The cells were incubated for 24 h at 37°C in 5% CO2 in a humidified cell culture incubator, then the medium was aspirated. The cells were washed twice with PBS and incubated for an additional 3 h in the medium supplemented with NR (50 µg/mL). After the medium was discarded, the cells were rinsed five times with warm PBS (pH 7.4) to remove the nonincorporated excess dye and 200 µL of destain solution (50% ethanol, 1% acetic acid, and 49% distilled water) was added to each well to fix the cells and bring the NR into solution. The plates were shaken for 20 min, and the absorbance of the solution in each well was measured in a microplate reader at 540 nm and compared with wells containing untreated cells. Results were expressed as the mean percentage of cell growth from three independent experiments.
Statistical analysis
Statistical analysis of numerical results obtained in cell experiments was performed using IBM SPSS Statistics version 22 software (IBM software, USA). All quantitative results were statistically analyzed by Student-Newman-Keuls (SNK) test to determine the importance of the difference between different groups. The significant value level is defined as p < 0.05.
Results and discussion
Morphology of electrospun nanofibers
The surface morphology of resulting electrospun fibers was investigated with SEM analysis. The SEM images and fiber diameter histogram data are shown in Figure 2(a) to (f) and Figure 3(a) to (f). Figure 3(a), (c), (e) show PVA, PVA-Gel and PVA-Alg fibers were long and continuous with the average diameters of 74.5 ± 15.4, 101.07 ± 29.9 and 269.51 ± 70.25 nm, respectively. Besides, all neat fibers except PVA nanofiber had smooth surfaces with non-beaded morphology. When FA was sprayed into the PVA nanofiber, average fiber diameter increased from 74.5 ± 15.4 to 291 ± 66.9 nm (Figure 3(a) and (b)). PVA/FA fibers showed smooth structure with deposited FA clusters onto fiber. Unfortunately, FA clusters were not seen too much on the surface of the PVA/FA fibers. This is due to the use of same solvent in both electrohydrodynamic process and thus FA clusters had been incorporated into the fiber structure.

SEM images of (a) PVA, (b) PVA/FA, (c) PVA-Gel, (d) PVA-Gel/FA, (e) PVA-Alg and (f) PVA-Alg/FA nanofibers (3kX).

The average fiber diameters of (a) PVA, (b) PVA/FA, (c) PVA-Gel, (d) PVA-Gel/FA, (e) PVA-Alg and (f) PVA-Alg/FA fibers.
After adding gelatin into PVA solution, thin and smooth with bead-free structure were occurred (Figure 2(c)). In the literature, it has been indicated that thinner fibers are formed by increasing electrical forces [50] and decreasing tip-to-collector distance [51]. Even though the applied voltage of the PVA-Gel fibers was higher than PVA fibers, the average diameter of PVA-Gel fibers was greater than PVA fibers (Figure 2(a) and (c), Figure 3(a) and (c)). This is related to effect of solvent volatility on fiber diameter [52]. The diameter of PVA-Gel/FA fibers also had a fairly even distribution ranging from60 to 220 nm (Figure 3(d)). The fibers showed bimodal size distribution due to the lack of homogeneity of fiber structure. This may have been caused by phase separation between blend mixtures. The resulting natural-synthetic fibers have non-homogeneous structure [53]. FA clusters caused formation of fiber structure and destroyed the homogenous fiber morphology, as well. Therefore, 2D flat fibers were formed in some regions. The diameter of fiber increased with addition of alginate to the PVA solution (Figure 2(e) and (f) and Figure 3(e) and (f)). Although there were FA clusters on the PVA-Alg fibers, the structure destroyed seriously during electrospraying of FA. The reason of this is similar to PVA/FA fibers related to same solvent for two hydrodynamic process.
As shown in Figure 3(e) and (f), nano-scale PVA-Alg and PVA-Alg/FA fibers were produced with diameters ranging from 150 to 550 nm and 50 to 350 nm, respectively. As a result, SEM images revealed FA dispersions have both partially deposited on the surfaces of hydrophilic fibers and incorporated into the fiber structure. When it is compared hybrid fibers and folic acid morphology, folic acid on aluminum foil and folic acid dispersion on surface of the fibers have similar structure (Figure S2). The average size of electrosprayed folic acid particles is 0.283 ± 0.01 µm (The maximum particle size is around 1.5 ± 0.07 µm while minimum one is 0.045 µm).
The pore size distribution histograms and mean pore size values were given in Figure S3. The porosity of nanofibers effects drug release behaviour of nanofibers. The porous architecture provides to increase of drug release percentage [54]. PVA-Gel/FA electrospun fibers have the largest pores among all fibers. It was observed the individual fibers resulting in a higher porosity and mean pore size compared to the FA loaded ones (Table 2).
Mean pore size and porosity for fibers.

Thermal properties
Figure 4(a) and (b) illustrates TGA and DTG thermograms for neat PVA, PVA blends and FA-loaded electrospun PVA and PVA blend fibers. All electrospun fibers showed weight loss in the range of 30–100°C related to moisture out of fibers. The amount of weight loss was different for all fibers. At this range (30–100°C), the maximum weight loss was seen for PVA-Gel/FA and the minimum was seen for PVA/FA mixture. The amount of the weight losses of fibers were 9.28% and 4.15% for PVA-Gel/FA and PVA/FA, respectively. Apart from the weight loss related to moisture out three steps of weight loss were observed for the neat PVA fiber. The major weight losses of 43% taken place in the temperature range from 210°C to 350°C due to dehydration of PVA [55,56]. The second step is dominated by chain scission with weight loss of 8.18%about 450°C to 600°C [57]. The last step was related to burning of the pyrolysis product which was formed during analysis in the N2 atmosphere. The weight loss of 3.77% of the neat PVA fibers was seen in the last step. It was found that FA-loaded PVA fibers started to decompose around 150°C with 45% weight loss and continue to 300°C. The following weight loss of 13% from 350°C to 460°C was attributed to both elimination reaction of PVA and loss of pterin and then p-aminobenzoic acid units in folic acid. The weight loss of 8.83% in the last step was related to burning of the pyrolysis product which was formed during analysis in the N2 atmosphere.
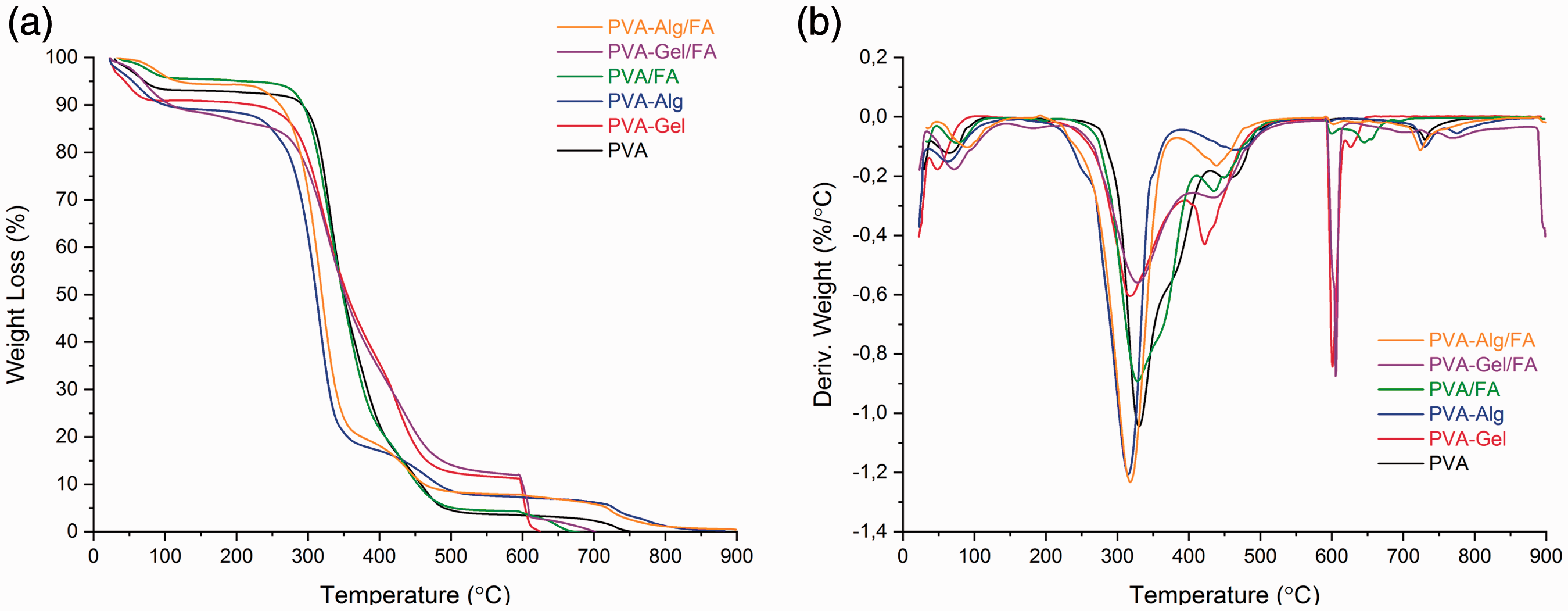
(a) TGA and (b) DTG thermograms of nanofibers.
The first weight loss of almost 67% apart from moisture out was seen between 280 and 450°C for PVA-Gel fibers. The second weight loss in the amount of 6.49% was occurred in the region of 450–550°C. The last weight loss was about 11.68% related to pyrolysis product which was formed during analysis in the N2atmosphere.The weight loss of blend fiber had a decrease at 280°C and continue to 500°C with 78% weight loss which was mainly due to the cleavage of –C=N bond is concerned with the presence of protein molecules in gelatin [58].
Thermal behavior of the PVA-Gel/FA was slightly difference from the thermogram of the PVA-Gel. The difference was only at the region of the 150–250°C temperature. There was slightly weight loss of the PVA-Gel/FA (about 3.34%). The other difference was occurred after the temperature of the 450°C due to the FA content. The decomposition speed was decreased as a consequence of –C=N group.
The lowest decomposition temperature was occurred for PVA/Alg blend nanofibers about 170°C [59]. The first decomposition weight loss of 68% from the fiber is related to dehydration of PVA. The second decomposition step temperature range of about 350–450°C, corresponding to the degradation of alginate with weight loss of 7.52%. The third decomposition step the temperature ranges of about 450–600°C with weight loss of 6.21% from the fiber was related to PVA chain scission as the other PVA samples. The loss of the moisture from PVA-Alg/FA was less than the PVA-Alg. Furthermore, as seen in Figure 4, PVA-Alg fibers showed faster decomposition than PVA-Alg/FA after 450°C. There is no residue for all NFs.
FT-IR analysis
The chemical composition of the all electrospun fibers and folic acid were determined through FT-IR analysis. As seen in Figure 5, the spectrum of folic acid has a number of characteristic peaks at 3590, 3496, 3330, 2925, 2840, 1694, 1650, 1605, 1487 and 1405 cm−1. The band between 3600–3300 cm−1 is associated with (–OH) stretching bands of glutamic acid moiety and –NH group of pterin ring. The band at 1650 cm−1 belongs to the (–C=O) stretching of (–CONH2). The another characteristic IR absorption peaks at 1605, 1694 and 1487 cm−1 is due to the N-H bending vibration of (–CONH) group, (–C=O) amide stretching of the α-carboxyl group and absorption band of phenyl ring respectively [60–62]. The frequencies for the neat PVA are indicated as follows: 3284 cm−1 for the stretching vibration peak of its (–OH) groups, 2943 and 2910 cm−1for the stretching vibration of –CH2group, 1242, 1088, 1023 and 945 cm−1 (C–O) stretching vibration, respectively [63,64]. On the other hand, for the PVA-Gel fibers the band appears at 3290 cm−1 for the (–OH) group. The bands appear at 2938 and 2913 cm−1 belongs to –CH2 stretching vibration. The characteristic absorption of gelatin peaks show mainly to the peptide bonds (–CONH) with the amide I–III vibrations. The peak is 1643 cm−1 (amide-I) is related to –C=O stretching vibration whereas the peak at 1535 cm−1 (amide-II) is due to N-H bending and C-H stretching vibration. 1243 cm−1 for the (amide-III) peak was occurred. In addition, 1435 cm−1 (–CH2 bending), 1374 cm−1 (C–H wagging), 1088 cm−1 (–C–O–C) and 837 cm−1 (C–C) stretching [64,65].

FT-IR spectra of nanofibers.
In literature, the characteristic peaks of alginate assigned 3297 cm−1 (–OH stretching), 2940 cm−1 (–CH2 stretching), 1612 and 1424 cm−1 asymmetric and symmetric (–COO) stretching and also 1096 cm−1 (C–O) stretching vibrations, respectively [66]. The electrospun PVA-Alg and PVA fibers were same absorption peaks approximately. However, the absorption peaks at 3284 cm−1 (–OH stretching) belongs to PVA that shifted to the 3297 cm−1 [23,67].
It is worth mentioning that the spectrums of NFs with drug (Figure 5) were similar the spectrum to neat NFs, indicating the absence of any chemical reaction between polymer and drug. This is explained by –OH groups in polymer structures shielded the characteristic peaks of drug molecules between 3300 and 3600 cm−1, corresponding to penetration of drug into the fibers. Additionally, the characteristic peaks of drug molecules could not be seen after 1700 cm−1 due to the low intensity.
UV-Vis spectroscopy- in vitro study
UV-Vis spectrometry was used to evaluate the release profile of FA from the nanofibers, which helped to reveal the structure-function relationship of the electrospun FA-loaded fibers in artificial sweat solution (pH 5.44, 37°C) during the time course of 480 min. The cumulative FA release rate profiles, in the sweat solution FA from the fiber samples, are plotted in Figure 6.
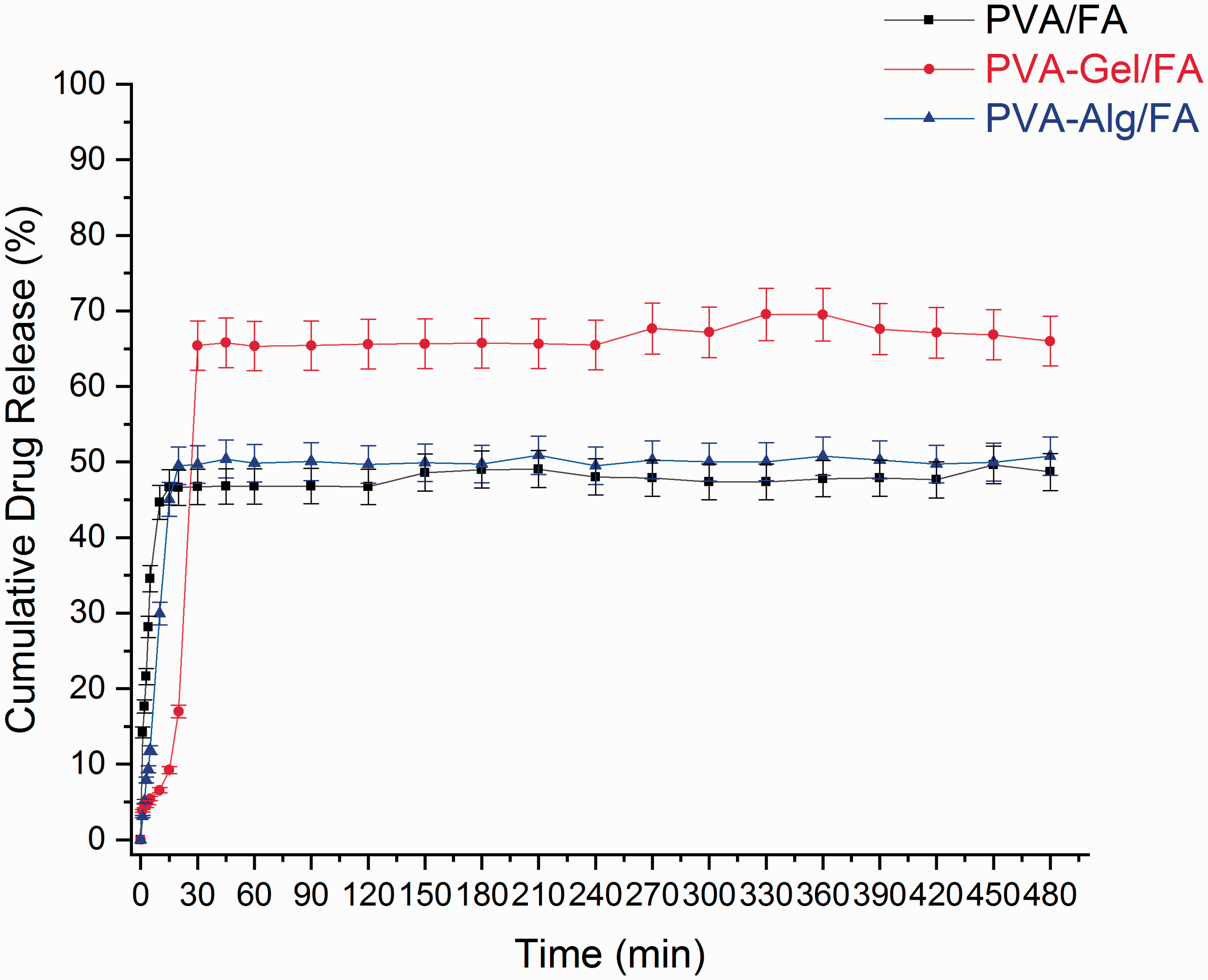
FA release profiles of nanofibers.
The release profile shows that the percentage cumulative FA releases are 49.6%, 69.55% and 50.88% for PVA/FA, PVA-Gel/FA and PVA-Alg/FA, respectively after 8 hours. As seen in Figure 6, all fibers exhibited initial burst release profile at an early stage of the analysis. Especially, in PVA/FA fiber at an early stage of the analysis within first 5 min, more than half of the FA was released from the fiber. In PVA-Gel/FA fiber within first 30 min about 65% FA release whereas in PVA-Alg/FA composite fibers 49% FA release. The rapid release of FA from the composite fibers is related to many factors, such as hydrophilic nature of matrix (PVA, Gel and Alg), high surface area of fibers that supports to increase in wettability [68,69]. Illangakoon et al. have reported hydrophilic PVP with paracetamol/caffein NFs release almost all caffein and paracetamol within the first 6 min [70]. In this study, the burst release behavior of all fibers might be related to the existence of hydrophilic based polymer matrix and drug as well. Furthermore, drug molecules deposited on the fiber surface also cause the initial release due to the hybrid process. On the other hand, burst release is a phenomenon which may be preferred to obtain quick results in dermal applications [71]. Kataria et al. also prepared ciprofloxacin loaded transdermal patches with fast release. In this study, the resulting PVA and PVA-Alg with ciprofloxacin fibers have fast release profile and reached maximum drug release curve [22].
It is clearly seen PVA-Gel/FA had the slowest behavior in fibers for 30 min. From the SEM images, the average fiber diameter of PVA/FA, PVA-Gel/FA and PVA-Alg/FA are 291±66.9, 145.33 ± 31.65 and 158 ± 48.55 nm. Moreover, the sample has the highest drug release rate compared with the other samples in total time. This is explained as the thickness of the fibers increase, the pathway drug diffusion of will increase [72].
The maximum drug was released from the fibers within sustained behaviour until 45 min. From the Figure 6 showed diffusion based drug release occured for 30 min, then followed by a constant drug release till 8 h due to erosion of polymer matrix. Similar UV-Vis results were reported by Arthani et al. [23]. Parın and Yıldırım also found burst release of folic acid (FA) from gelatin fibers for 30 min [73]. However, the release rate in PVA-Gel/FA fiber is partially slower than others for the first 30 min. due to the interaction hydrogen bonding between PVA, gelatin and folic acid. PVA-Alg/FA fiber also indicated similar situation, but unfortunately the low amount of alginate in the fiber limit the formation of too many interaction of hydrogen bonding (Figure 5). Consequently, PVA-Gel/FA could be used as an efficient drug delivery system for beauty mask purpose with 8 hours-period.
Entrapment efficiency (EE) and loading capacity
The drug solubility in electrospun fibers effects entrapment efficiency and loading capacity of FA-containing nanofibers [25]. These two values were calculated by UV-Vis spectrophotometer due to the nonvolatile structure of FA.
The resulting FA loading capacity and the entrapment efficiencies of the fibers are shown in Table 3.
The entrapment efficiency and loading capacities of fibers.

Cytotoxic effects of nanofibers by MTT assay
The cytotoxic effects of PVA/FA, PVA-Gel/FA and PVA-Alg/FA nanofibers extract solutions and also pure folic acid (2200 µg/mL) concentration were evaluated by MTT assay in L929 cells. To determine and compare the cytotoxicity, extract solutions of all NFs and also a negative control group without any chemicals, aluminum foil as a planch material and a positive control group containing 1% Triton x-100 were prepared. Eight wells of each sample were repeated in 96 well plates. The average absorbance values and standard deviation values of living cells were calculated by averaging all the data obtained. In addition, cell viability in the control group was considered as 100%, and living cell percentages were determined for all samples compared to the control. Results of MTT test are shown in Figure 7. According to the results of MTT test applied after 24 h of treatment in L929 cells, the groups treated with nanofiber extracts caused statistically significant (p < .0.05) decreases in cell viability in all groups compared to the negative control group. However, since viability did not fall below 70%, it was not evaluated as cytotoxic by MTT assay. Folic acid did not cause any cytotoxic effects in and even caused increases in cell proliferation compared to negative control. This situation also prove that folic acid plays a crucial role cell growth, and development, synthesis of nucleic acids and metabolism of many amino acids.

Effects of PVA nanofiber extract solutions on cell viability of L929 cells by MTT and NRU assays. Bars that do not share same numbers or letters (superscripts) are significantly different from each other (p < 0.05).
According to SNK analysis all samples were between positive control (1) and FA (5). FA increase cell proliferation instead of decrease. Because of that cell viability was more than the negative control. PVA-Gel and PVA-Alg were in the same group (2). PVA-Alg also place in 3rd group. PVA, PVA-Gel/FA and PVA/FA were in the same group (both 3 and 4) with negative control. PVA-Alg/FA placed in 3 group. According to SNK analysis PVA-Gel-FA, PVA and PVA-FA samples were more effective respectively to cell viability than the others. According to MTT analysis test results, all samples could be acceptable as biocompatible according to ISO 10993-5.
Cytotoxic effects of nanofibers by NRU assay
The cytotoxic effect of the extract solutions of prepared NFs were evaluated by NRU assay in L929 cells. Results of NRU test are shown in Figure 7. According to the results of NRU test applied after 24 h of treatment in L929 cells, the groups treated with nanofiber extracts caused statistically significant (p < .0.05) decreases in cell viability in all groups compared to the negative control group. Except the PVA-Gel NF cell viability decreased under 70%, but not decreased under 50%. PVA-Alg NFs caused more cytotoxicity in NRU assay. On the other hand, L929 cells were treated with a high concentrations of nanofiber extracts in this study to investigate exact cytotoxicity.
In the NRU method, which is based on the measurement principle of lysosomal activity, cell viability is shown by a different mechanism than MTT assay (based on the measurement of mitochondrial activity as metabolic activity). It is thought that the nanofiber extract solutions may cause toxic effects by interacting with the cell membrane or enter the cell through the membrane and accumulating in organelles and cytoplasm.
SNK analysis showed that all samples were between positive control (a) and FA (e). PVA-Alg, PVA-Alg/FA, PVA/FA and PVA were place in the same group (b). PVA and PVA-Gel/FA also formed a new group between the other groups (c). PVA-Gel was a single group in itself and place with negative control (d). The cell viability was about similar with negative control samples. PVA-Gel was more effective to cell viability than the others. According to NRU analysis test results, PVA-Gel and PVA-Gel/FA could be acceptable as biocompatible according to ISO 10993-5.
In this study nanofiber samples were sterilizied with ultraviolet (UV) radiation for in vitro cytotoxicity assessments. Although UV is considered a disinfection method not a sterilization method, it is generally used in laboratory-scale noncontact sterilization processes. Microorganisms especially bacteria are inactivated when exposed directly to UV radiation. Its maximum bactericidal effect occurs at 240–280 nm and depends of time of exposure [74]. We sterilized each side of the nanofibers under a UV lamp (254 nm) for 1 h in a laminar flow cabinet (Telstar BioVanguard Class II Biological Safety Cabinet). On the other hand, as discussed recently in the literature although standard sterilization procedures are available for many materials today, there is no standard procedure for materials prepared from nanofibers. It is well known that the sterilization process should be selected carefully because it may affect the performance of scaffold [75]. It was indicated that the use of UV for sterilization of nanofibers was found to be the least damaging sterilization method on nanofiber morphology, mechanical properties, and water contact angle [76,77]. Tipnis and Burgess (2018) suggested that UV sterilization can be used for temperature and moist sensitive medical devices and pharmaceutical products [78]. Rediguieri et al. (2016) reported the impact of sterilization methods on electrospun scaffolds for tissue engineering and indicated that the effects of sterilization could have different out comes due to various properties of electrospun fibers, such as high porosity and surface area [79]. Known different sterilization methods for nanofibers area utoclave, dryheat, gamma radiation, electron beam radiation, ethyleneoxide, peracetic acid (paa), plasma and ozone, disinfection methods are ultraviolet radiation and ethanol solutions. In this study we did not compare the sterilization methods on material properties and characterization tests did not done after UV exposure, but research on this issue is important for clinical use.
Conclusion
In this study, we successfully produced fast-dissolving drug delivery systems derived from hydrophilic composite fibers by simultaneous electrospraying and electrospinning method. FA was used as model drug and the resulting composite fibers consists of FA clusters entrapped in PVA, PVA-Gel and PVA-Alg NFs. The SEM images showed the composite fibers possessed relatively uniform with average fiber diameters between 145 and 291 nm and most of the FA clusters have been integrated into the fibers rather than deposit on the fiber surface due to polymers and folic acid solutions are hydrophilic based nature. FTIR spectra results demonstrated the apparent slight shifts of some peaks, corresponding to physical interactions of FA with fibers. The TGA results pointed out that by the incorporation of FA into NFs, the degradation rate was increased slightly. However, FA was stable at high temperature. The in-vitro release test clearly confirmed that the obtained PVA/FA, PVA-Gel/FA and PVA-Alg/FA NFs could release the FA in a sustained manner with initial burst release for the 8 hour-period. The observation of fast dissolving of all fibrous structures with FA in 30 min is directly related to two reasons: the strong hydrophilic nature of PVA, PVA-Gel and PVA-Alg NFs and FA clusters deposited on fiber surfaces due to electrospraying process.
The cytocompatibility test results based on the cytoxicity methods adapted from the ISO10993-5 standards indicated novel PVA/FA, PVA-Gel/FA and PVA-Alg/FA electrospun nanofibrous patches revealed no cell toxicity on cultured fibroblasts in MTT assay but slight cell toxicity in NRU assay. In this study L929 cells were treated with a high concentrations of nanofiber extracts to investigate exact cytotoxicity due to the nature of certain biodegradable/leachable and extractable biomaterials. As indicated in cytocompatibility test protocols the quantity exposed to used cell lines is dependent to the interface area, the volume of extraction, pH, temperature, time and many other factors. Results of this study and similar studies in the literature also indicated that the cytotoxic effect the different PVA nanofibers are based on the components and content amounts. Our results reveal that PVA and PVA-Gel with/without FA nanofibers seems more biocompatible than PVA-Alg nanofibers. However, further cytocompatibility tests should be carried out in different conditions, concentrations and different cell lines according to potential use for dermal or other biomedical applications as scaffolds.
Recently, there have been a variety approaches about
Supplemental Material
sj-pdf-1-jit-10.1177_1528083721997185 - Supplemental material for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility
Supplemental material, sj-pdf-1-jit-10.1177_1528083721997185 for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility by Fatma Nur Çiğdem İnci Aydemir, Gökçe Taner and Kenan Yıldırım in Journal of Industrial Textiles
Supplemental Material
sj-pdf-2-jit-10.1177_1528083721997185 - Supplemental material for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility
Supplemental material, sj-pdf-2-jit-10.1177_1528083721997185 for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility by Fatma Nur Çiğdem İnci Aydemir, Gökçe Taner and Kenan Yıldırım in Journal of Industrial Textiles
Supplemental Material
sj-pdf-3-jit-10.1177_1528083721997185 - Supplemental material for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility
Supplemental material, sj-pdf-3-jit-10.1177_1528083721997185 for Co-electrospun-electrosprayed PVA/folic acid nanofibers for transdermal drug delivery: Preparation, characterization, and in vitro cytocompatibility by Fatma Nur Çiğdem İnci Aydemir, Gökçe Taner and Kenan Yıldırım in Journal of Industrial Textiles
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Bursa Technical University Scientific Research Projects Coordination Unit (SRP Project No. 190D003). We would like to thank the employees of the Scientific and Technological Research Council of Turkey- Bursa Test and Analysis Laboratory (TUBİTAK-BUTAL) for supporting the preparation of artificial sweat solutions and thank to İbrahim ŞEN (Central Research Laboratory, Bursa Technical University) for his help in TGA analysis.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.