
Abstract
Diabetic cardiomyopathy is a major contributor to the increasing burden of heart failure globally. Effective therapies remain elusive, in part due to the incomplete understanding of the mechanisms underlying diabetes-induced myocardial injury. The objective of this study was to assess the direct impact of insulin replacement on left ventricle structure and function in a rat model of diabetes. Male Sprague-Dawley rats were administered streptozotocin (55 mg/kg i.v.) or citrate vehicle and were followed for 8 weeks. A subset of diabetic rats were allocated to insulin replacement (6 IU/day insulin s.c.) for the final 4 weeks of the 8-week time period. Diabetes induced the characteristic systemic complications of diabetes (hyperglycaemia, polyuria, kidney hypertrophy) and was accompanied by marked left ventricle remodelling (cardiomyocyte hypertrophy, left ventricle collagen content) and diastolic dysfunction (transmitral E/A, left ventricle-dP/dt). Importantly, these systemic and cardiac impairments were ameliorated markedly following insulin replacement, and moreover, markers of the diabetic cardiomyopathy phenotype were significantly correlated with the extent of hyperglycaemia. In summary, these data suggest that poor glucose control directly contributes towards the underlying features of experimental diabetic cardiomyopathy, at least in the early stages, and that adequate replacement ameliorates this.
Keywords
Introduction
The global incidence of diabetes is predicted to reach 642 million adults by 2040, impacting both human health and world economies.1,2 Heart failure is increasingly recognised as a major contributor to morbidity and mortality in diabetic patients.3,4 Not only do diabetics suffer increasing incidence of heart failure, their prognosis once diagnosed is worse than a comparable non-diabetic patient.3,4 This may partially be explained by the distinct form of heart failure present in diabetic patients, termed diabetic cardiomyopathy, that can develop independently of other comorbidities including hypertension, coronary heart disease or hyperlipidaemia,5–7 and currently has no specific treatment. Indeed, the impact of diabetic management strategies on diabetic cardiomyopathy is poorly understood.
Type 1 diabetes represents 5%–10% of global diabetic patients and is characterised by early onset of hyperglycaemia, although both type 1 and type 2 diabetes lead to severe cardiovascular complications, as well as functional and structural changes in the heart that make it more susceptible to stress.2,6 Indeed, a prominent feature of the diabetic heart is the development of diastolic dysfunction, now a well-recognised clinical entity, 8 with subclinical diastolic dysfunction occurring in up to 60% of optimally controlled diabetic patients. 9 Diabetes-induced contractile and relaxation abnormalities are evident in isolated ventricular myocytes in vitro as early as 4–6 days after the induction of streptozotocin (STZ)-induced diabetes in rats in vivo 10 and are readily replicated by incubation under high glucose conditions in vivo. 11 The understanding of the cellular and molecular perturbations that predispose to altered myocardial structure is lacking, partially due to the chronic and often asymptomatic nature of diabetic cardiomyopathy, even in patients with adequate metabolic control, resulting in patients going undiagnosed until more acute complications develop. 12
The initial manifestations of diabetic cardiomyopathy include diastolic stiffness, abnormalities further exacerbated following ischaemia, followed by systolic and autonomic dysfunction in more advanced cases.13,14 Advances in this field have implicated several processes in the pathogenesis of diabetic cardiomyopathy beyond myocardial hypertrophy, myocardial fibrosis, metabolic disturbances, small vessel disease and cardiac autonomic neuropathy.6,13,15,16 These pathways are currently the subject of intense research to develop therapeutics that directly prevent or delay the progression of heart failure in diabetic patients and have been reviewed extensively elsewhere.6,13
The current therapeutic strategies for treating heart failure in diabetics and non-diabetics remain the same, despite diabetes being an independent predictor of poor prognosis of heart failure in clinical trials. This suggests that diabetic cardiomyopathy is driven by an increase in glucose levels and impairments in glucose control. However, even though the epidemiological link between plasma glucose levels and adverse cardiovascular events is well established, with a 1% increase in HbA1c conferring an 8% increased risk of developing heart failure, 17 large prospective trials have provided conflicting results regarding intensive glucose control as a possible intervention.18–20 Therefore, given the conflicting data on optimising glycaemic control in diabetic patients, this study sought to investigate the apparent contribution of insulin deficiency, and the subsequent reduction in blood glucose levels after insulin replacement, on the development of severe experimental diabetic cardiomyopathy, in an experimental model of STZ-induced type 1 diabetic cardiomyopathy.
Methods
Induction of diabetes
Eight-week-old male Sprague-Dawley rats (229 ± 5 g) were administered either STZ (55 mg/kg via the tail vein, to induce type 1 diabetes) or citrate buffer vehicle (0.42% in sterile saline, pH 4.5, non-diabetic controls) after an overnight fast as previously described (Supplementary Figure 1). 21 Diabetic rats were given sucrose-supplemented (15 g/L) drinking water for the initial 48 h after STZ (to limit early mortality), after which they were given normal drinking water ad libitum. After 4–5 days, urinalysis confirmed successful induction of diabetes (Clinistix Reagent Strips, Bayer Pymble Australia). After 4 weeks, a subset of diabetic rats were allocated to insulin replenishment, via long-acting human Ultralente insulin (6 IU/day Ultratard HM; Novo Industries, Bagsvaerd, Denmark). The remaining diabetic and non-diabetic animals remained untreated. Rats were then followed for an additional 4 weeks (i.e. a total study period of 8 weeks). At study endpoint, non-fasting blood glucose was determined via handheld glucometer. As the upper limit of detection for blood glucose readings was 33.3 mM, ‘High’ readings were recorded as 33.3 mM. All animal research was conducted in accordance with the ‘National Health and Medical Research Council of Australia’ guidelines and by the Animal Ethics Committees of the Howard Florey Institute and the Alfred Medical Research and Education Precinct (AMREP).
Haemodynamic and cardiac function measurements in vivo
Systolic blood pressure (via tail-cuff plethysmography in conscious rats 22 ) and 24 h urine volume (via metabolic caging) were assessed in the final week of diabetes or sham. At study endpoint (after 8 weeks of diabetes or sham), cardiac function was assessed in anesthetised rats (ketamine/xylazine/atropine: 60/12/0.6 mg/kg i.p.) via echocardiography followed by cardiac catheterisation, as previously described. 23 Two-dimensional M-mode and Doppler flow echocardiography was performed, using a SONOS 5500 ultrasound machine and a 12-MHz probe, to assess left ventricle (LV) systolic and diastolic function at study endpoint, respectively. Arterial blood and LV pressures were also assessed by catheterisation using a 2Fr Millar MIKRO-TIP catheter and a PowerLab System (AD Instruments, Bella Vista, NSW, Australia), immediately after final echocardiography, while rats were still anesthetised. Parameters of LV diastolic function included LV filling, assessed on the peak E and A wave blood flow velocities and their ratio, E/A ratio, in addition to LV-dP/dt (the minimum rate of LV pressure change). LV systolic function was assessed on LV dimensions derived from M-Mode echocardiography, peak aortic flow velocity (Doppler echocardiography) and both LV systolic pressure and LV + dP/dt (the maximum rate of LV pressure change), as described previously. 23
Tissue collection
Immediately following cardiac catheterisation, while still anesthetised, rats were euthanised by exsanguination, and hearts and kidneys were collected. A portion of LV was cut, fixed in 10% neutral buffered formalin (Australian Biostain, Melbourne, VIC, Australia) and paraffin-embedded for histological analysis. 24 Small portions of LV (~1 to 2 mm in diameter) were kept aside for estimation of superoxide levels in fresh tissue,25,26 and the remainder of the LV was snap-frozen in liquid nitrogen and stored at −80°C for biochemical analysis.24,25
Histology
Paraffin-embedded LV cross sections were cut (4 µm) and stained with hematoxylin and eosin (H&E) or 0.1% picrosirius red and analysed as previously described. 24 H&E-stained sections were photographed under light microscopy at ×400 magnification (with resolution of 2048 × 1536 pixels and 300 × 300 dpi), and cardiomyocyte width manually determined using Olympus Image Pro-plus (Media Cybergenetics, Bethesda, MD 24 ). Cardiac collagen deposition was also assessed on 0.1% picrosirius red-stained sections. In total, 10 fields were randomly chosen, taking care to avoid arterial collagen deposition, and photographed via light microscopy at ×200 magnification. LV interstitial collagen staining was also analysed using Olympus Image Pro-plus, as described.24,25
Analysis of LV NADPH oxidase activity
LV NADPH oxidase activity was assessed as superoxide levels, determined in fresh LV tissue via β-NADPH-driven (100 µM) lucigenin (5 µM)-enhanced chemiluminescence, as previously described.25,26 Tissues were then dried at 60°C for 48 h, for subsequent calculation of superoxide levels as relative light units (RLU) per second per milligram of tissue. Results were expressed as fold non-diabetic shams studied in parallel.
Analysis of LV gene expression
RNA was extracted from frozen LV using TRIzol® reagent (Invitrogen Life Technologies, Mount Waverley, VIC, Australia) as per the manufacturer’s instructions, and reverse-transcribed as previously described. 24 LV expression of the pro-hypertrophic markers β-myosin heavy chain, atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP), the p47phox and Nox2 subunits of NADPH oxidase, the pro-inflammatory markers tissue necrosis factor-α (TNF-α), monocyte chemoattractant protein-1 (MCP-1) and tribbles homolog-3 (TRB3), as well as the inflammatory regulator annexin-A1 and its formyl peptide receptors (FPRs), FPR1 and FPR2, and the housekeeping gene 18S, were determined via real-time polymerase chain reaction (PCR) using SYBR Green chemistry (Applied Biosystems, Scoresby, VIC, Australia), and primers generated from rat sequences in GenBank. Quantitative analysis was performed using ABI Prism® 7700 Sequence Detection software, using ΔΔCt method to detect fold differences relative to control group. 24
Statistical analysis
Data were analysed with GraphPad Prism 7.01 statistical software package. All data are presented as mean ± standard error (SE). One-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was used to analyse the differences between the three experimental groups: non-diabetic sham, untreated diabetes and insulin-treated diabetes. Correlations were also sought between blood glucose and LV diastolic function (on E/A ratio and LV-dP/dt) using simple linear regression analysis. All variables were normally distributed as tested with the D’Agostino-Pearson omnibus normality test. Statistical significance was considered at p < 0.05.
Results
Characterisation of the diabetic phenotype
Male Sprague-Dawley rats injected with STZ displayed elevated plasma glucose levels and were deemed to be diabetic (Supplementary Figure 1). Rats receiving daily injections of insulin in the last 4 weeks of the 8-week diabetic model exhibited significantly reduced plasma glucose levels (Figure 1(a)). Untreated diabetic rats failed to gain weight at the same rate as non-diabetic rats (Figure 1(b)), thus causing a reduced final body weight (Figure 1(c)), although insulin replacement resulted in an improvement in weight gain towards non-diabetic controls. Diabetic animals displayed polyuria, an effect significantly attenuated by insulin treatment (Figure 1(d)). The absolute weights of the right and left kidneys were the same in all groups (results not shown); however, when these weights were normalised to body weight, these values were significantly increased in diabetic rats (Figure 1(e) and (f)), an effect that was ameliorated by insulin replacement.
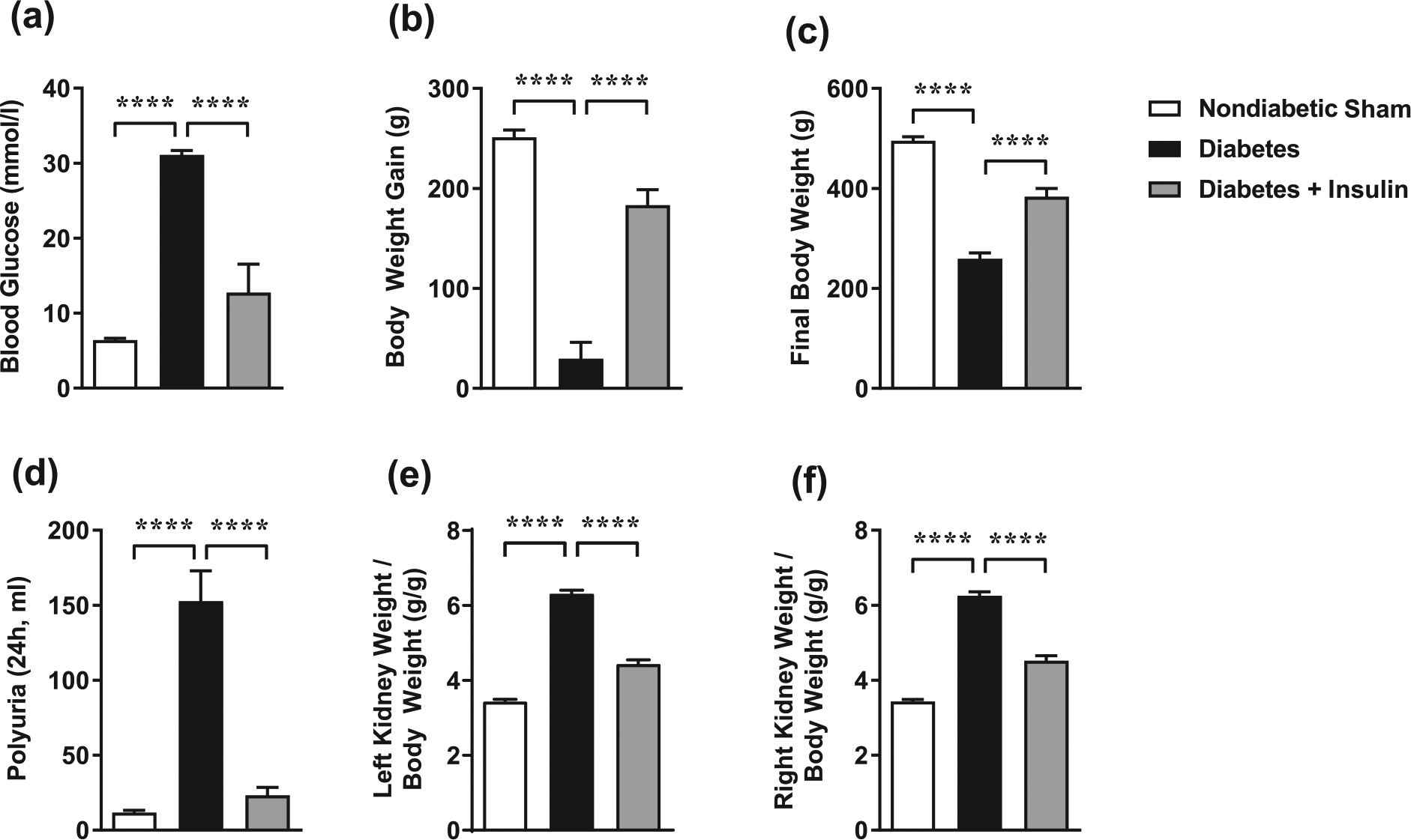
Insulin replacement for the final 4 weeks of 8-week STZ-induced diabetes ameliorates the systemic and morphological changes in rats. (a) Blood glucose levels, (b) body weight gain, (c) final body weight, (d) polyuria, (e) left kidney weight/body weight and (f) right kidney/body weight were partially protected from diabetes-induced impairments following insulin replacement. Values are means ± SE. n = 5–32 per group. ****p < 0.0001 (one-way ANOVA followed by Tukey’s post hoc test).
Cardiac dimensions, blood pressure and LV function
In conscious rats, tail-cuff measurements of systolic blood pressure were unchanged between all treatment groups (Table 1). In anaesthetised rats, both systolic and diastolic blood pressures were reduced in the diabetic group, a difference not observed in the insulin replacement group. Cardiac structure and function were assessed by echocardiography. Heart rate was reduced in diabetic animals compared to the non-diabetic group and changed following insulin replacement. The cardiac dimensions, LV end diastolic dimension (LVEDD) and LV end systolic dimension (LVESD) were reduced significantly in diabetic animals (Table 1), in the case of LVEDD insulin replacement attenuated this effect.
Conscious tail-cuff blood pressure, anaesthetised catheter blood pressure and LV functional parameters by echocardiography in anaesthetised STZ-induced diabetic rats.
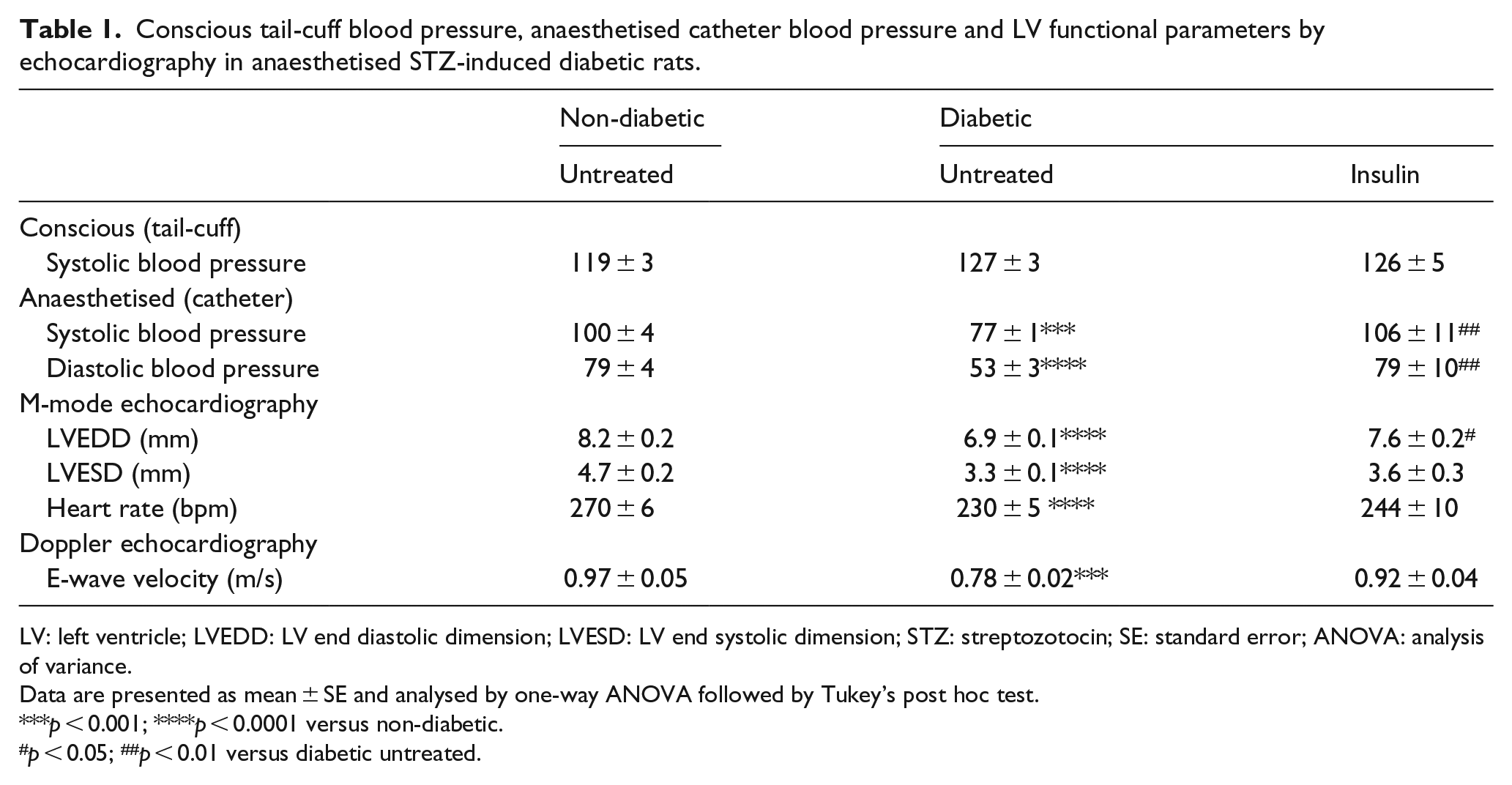
LV: left ventricle; LVEDD: LV end diastolic dimension; LVESD: LV end systolic dimension; STZ: streptozotocin; SE: standard error; ANOVA: analysis of variance.
Data are presented as mean ± SE and analysed by one-way ANOVA followed by Tukey’s post hoc test.
p < 0.001; ****p < 0.0001 versus non-diabetic.
p < 0.05; ##p < 0.01 versus diabetic untreated.
Measurements of LV function using a Millar catheter positioned in the LV revealed a reduction in LV –dP/dt in diabetic animals that was reversed by insulin treatment (Figure 2(a)). Doppler echocardiographic assessment of diastolic function revealed a significant reduction in E-wave velocity (Table 1) and a parallel increase in A-wave velocity following STZ-induced diabetes (Figure 2(b)). Both of these parameters were ameliorated by insulin replacement resulting in significant improvements in E/A ratio (Figure 2(c)). A-E time, another measure of LV relaxation, was increased in diabetic rats and was attenuated with insulin replacement (Figure 2(d)). A linear regression was calculated for markers of diastolic dysfunction, namely, E/A ratio (Figure 2(f)) and LV-dP/dt (Figure 2(g)) based on blood glucose levels. Interestingly, both E/A ratio (p < 0.0001) and LV-dP/dt (p < 0.0001) were significantly inversely correlated with blood glucose levels.
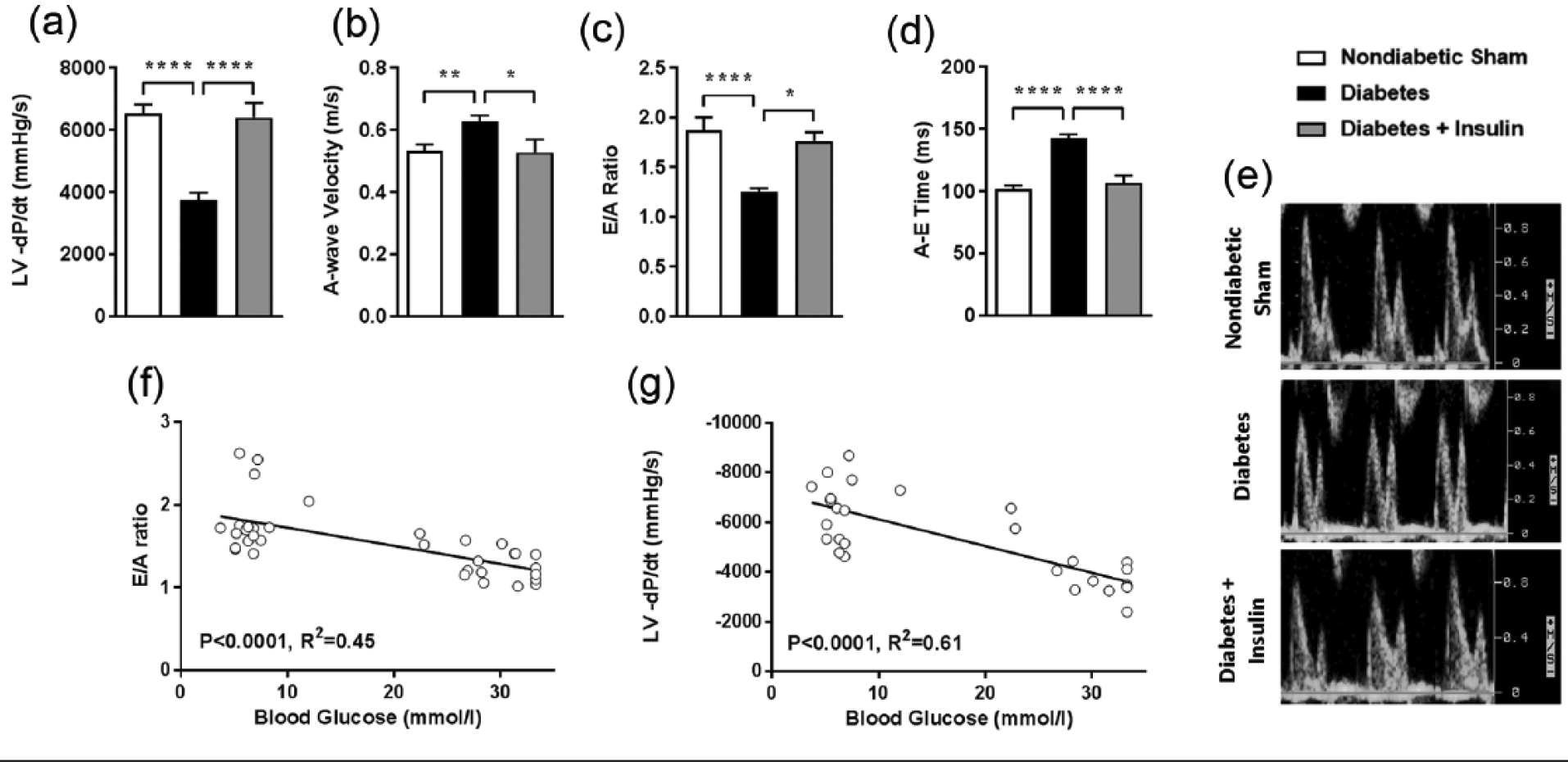
Insulin replacement for the final 4 weeks of 8-week diabetes ameliorates diabetes-induced diastolic dysfunction. Cardiac catheterisation was used to determine maximum rate of fall of LV pressure, (a) an index of LV relaxation (LV-dP/dt); Doppler echocardiography was used to derive peak (b) A-wave velocity, (c) E/A ratio and (d) A-E time, as depicted in the (e) representative images. Linear regression analysis was used to predict (f) E/A ratio and (g) LV-dP/dt based on blood glucose levels. Values in bar graphs are means ± SE. n = 5–20 per group. *p < 0.05; **p < 0.01; ****p < 0.0001 (one-way ANOVA followed by Tukey’s post hoc test).
LV external dimension was reduced by diabetes and attenuated by insulin replacement (Figure 3(a)). In terms of systolic function, LV posterior wall thickness (Figure 3(b)) was significantly reduced following STZ-induced diabetes, an effect reversed by insulin replacement. Similarly, peak aortic flow velocity (Figure 3(c)) and aortic ejection time (Figure 3(d)) were less efficient in diabetic rats, but were effectively restored following insulin replacement. Furthermore, diabetes reduced LVSP and LV + dP/dt, but both abnormalities were attenuated by insulin treatment (Figure 3(e) and (f)).
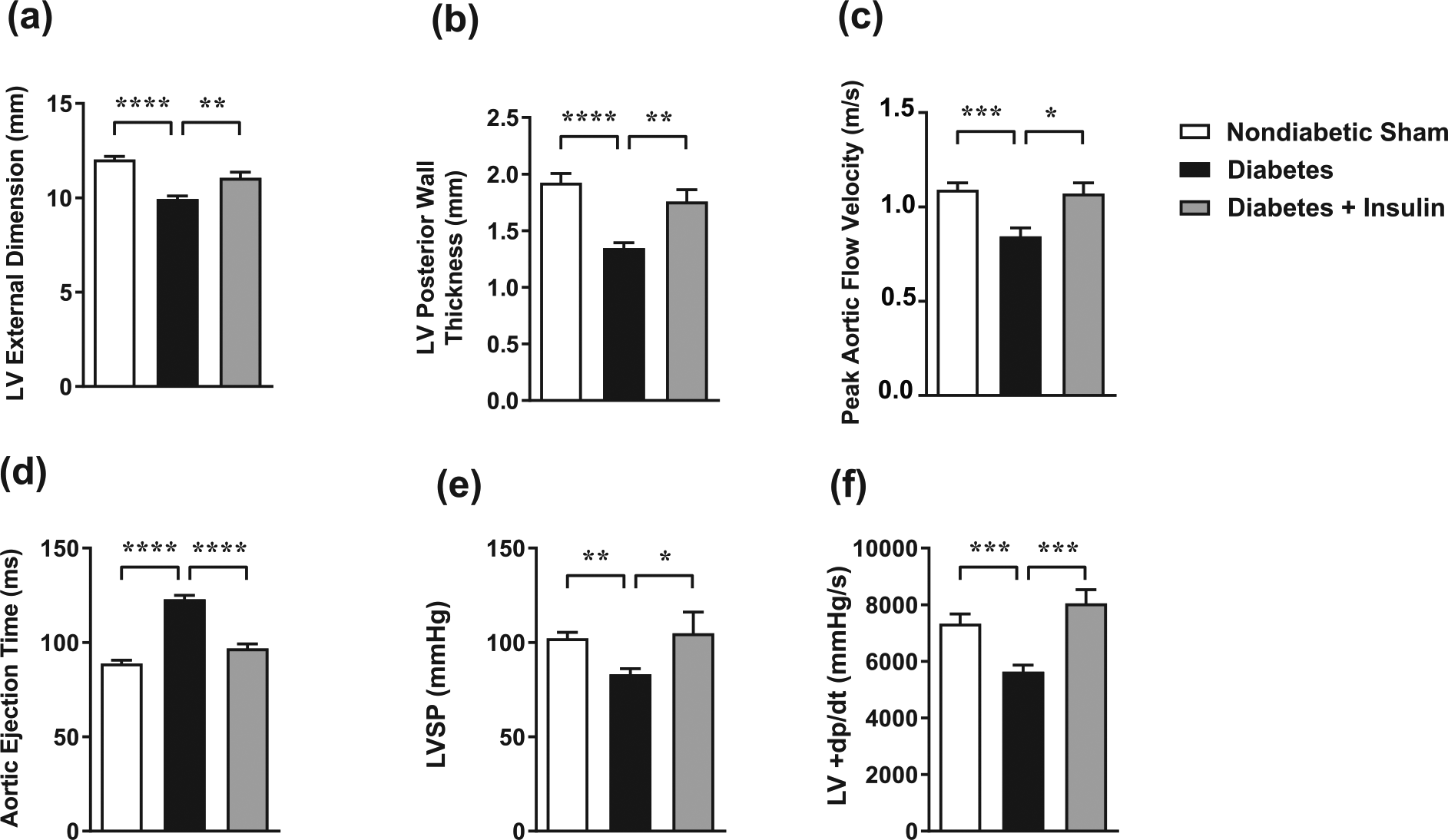
Insulin replacement for the final 4 weeks of 8-week diabetes ameliorates diabetes-induced systolic dysfunction. Echocardiography was used to calculate (a) LV external diameter, (b) LV posterior wall thickness, (c) peak aortic flow velocity and (d) aortic ejection time. Cardiac catheterisation was used to measure (e) LV systolic pressure (LVSP) and derive the maximum rate of rise of LV pressure, (f) an index of LV contractility (LV + dP/dt). Values are means ± SE. n = 6–15 per group. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (one-way ANOVA followed by Tukey’s post hoc test).
Effect of insulin treatment on diabetes-induced cardiac fibrosis and cardiomyocyte hypertrophy
In diabetes-induced heart failure, the myocardium undergoes a number of characteristic structural changes. One such change is the increased deposition of collagen in the LV. In this study, LV collagen deposition was markedly increased in diabetic rats compared to non-diabetic controls, an effect reversed by insulin administration (Figure 4(a) and (b)). Another common structural feature of diabetic cardiomyopathy is cardiomyocyte hypertrophy. In this study, diabetic animals displayed an increase in cardiomyocyte width that was effectively reversed by insulin treatment (Figure 4(c) and (d)). Furthermore, mRNA expression of selected markers of LV hypertrophy, including LV ANP, β-myosin heavy chain expression and BNP, were increased in the diabetic group (Figure 5(a)–(c)). Diabetic animals receiving insulin replacement did not display increased expression of these markers. mRNA expression of glucose transporter GLUT4 was significantly decreased in the diabetic rats and restored by insulin treatment (Figure 5(d)). Absolute heart weight was decreased in diabetic rats and while this effect was attenuated by insulin treatment (Figure 5(e)), it is likely an artefact of the characteristic changes in body weight in the STZ model. When heart weight was normalised to body weight, diabetic animals displayed a hypertrophic phenotype (Figure 5(f)). Insulin replacement reversed absolute heart size and final body weight, with no statistical difference in heart weight to body weight ratio.
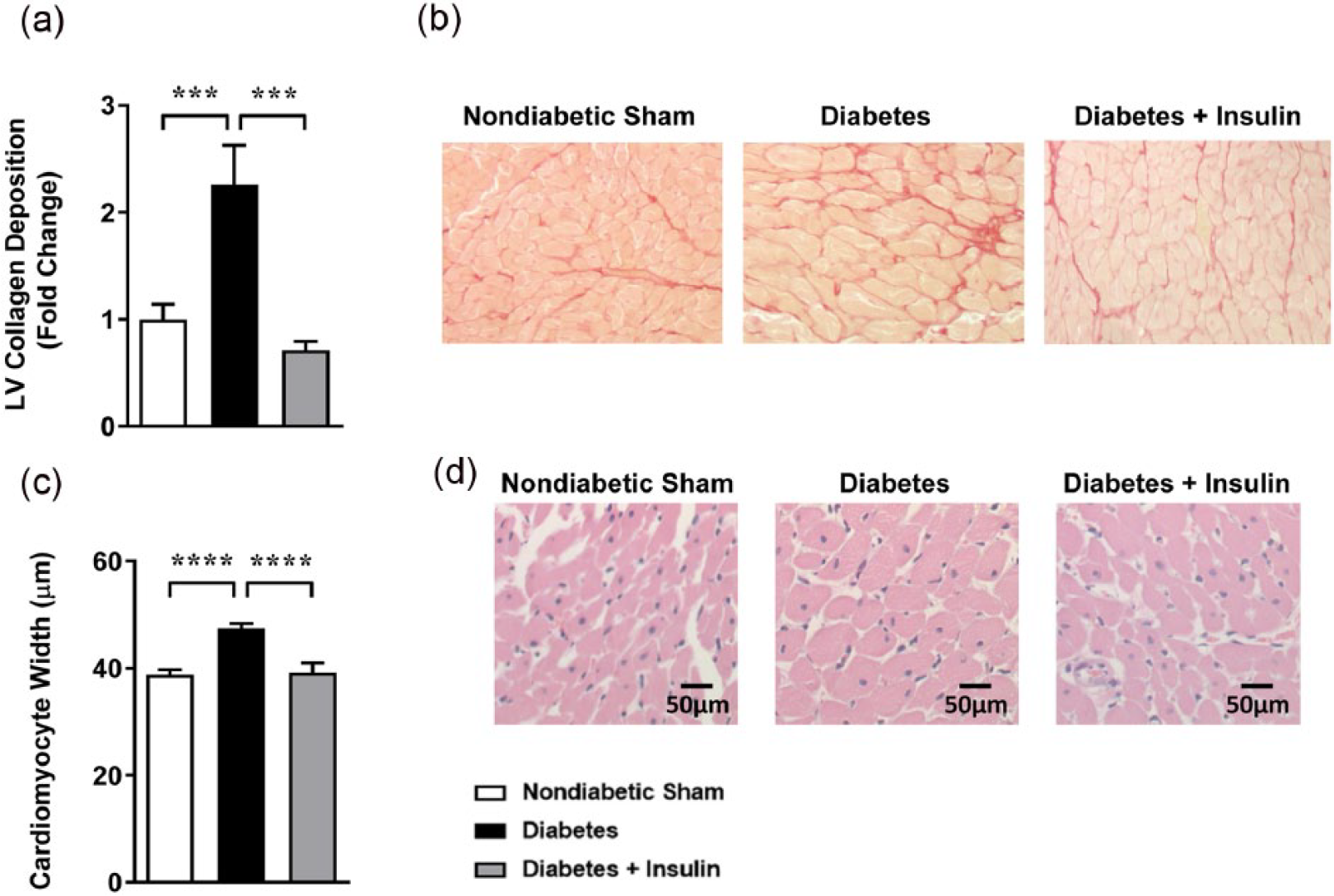
Insulin replacement for the final 4 weeks of 8-week STZ-induced diabetes ameliorates diabetes-induced cardiac structural changes. (a) Mean data and (b) representative images of collagen-stained LV cross sections with picrosirius red. (c) Mean data of cardiomyocyte width and (d) representative images (scale bar = 50 µm) from haematoxylin and eosin-stained LV sections. Values are means ± SE. n = 11–23 per group. ***p < 0.001, ****p < 0.0001 (one-way ANOVA followed by Tukey’s post-hoc test).
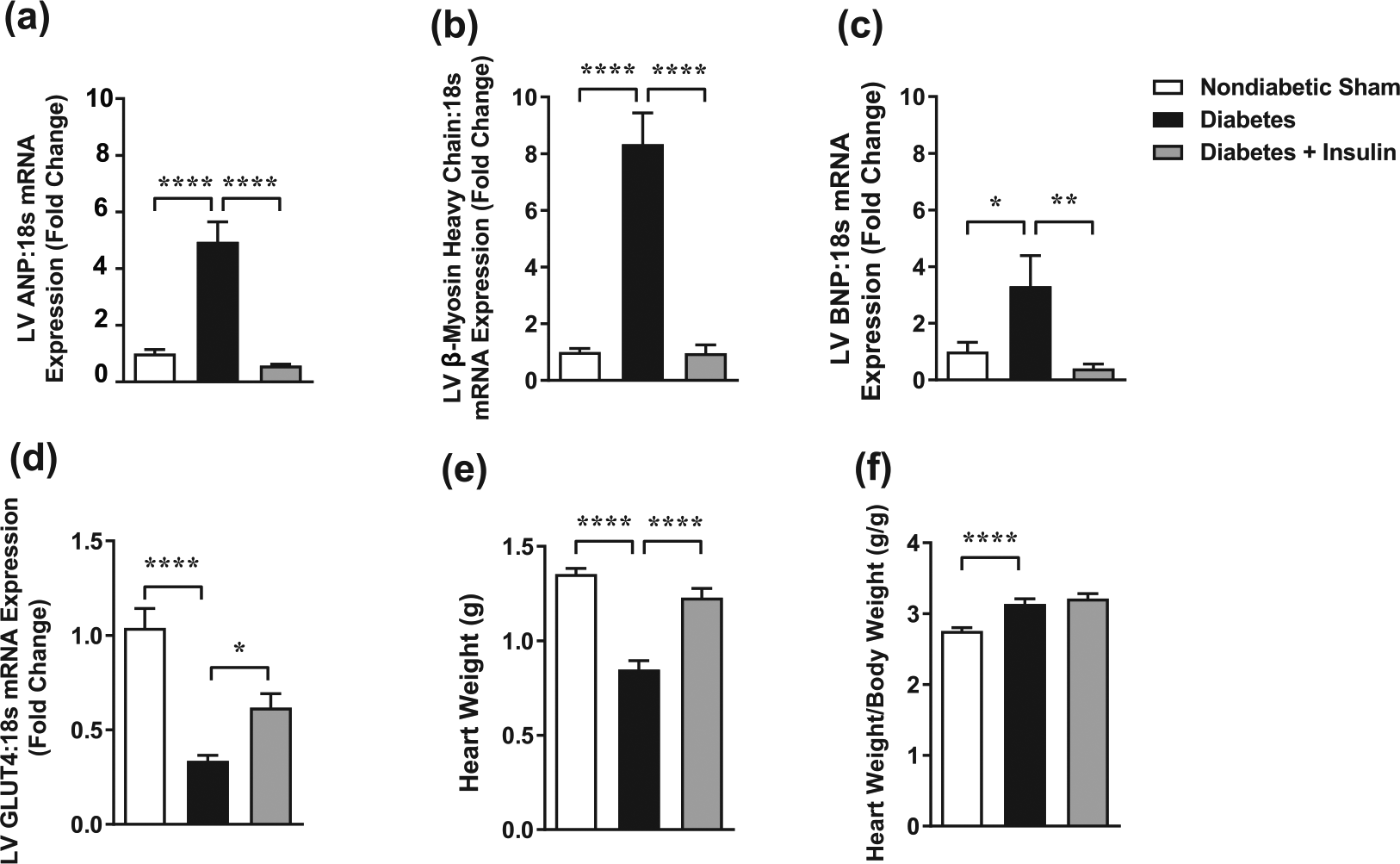
Insulin replacement for the final 4 weeks of 8-week STZ-induced diabetes ameliorates diabetes-induced cardiomyocyte hypertrophy. (a) Atrial natriuretic peptide (ANP), (b) β-myosin heavy chain and (c) B-type natriuretic peptide (BNP), markers of cardiomyocyte hypertrophy, and (d) glucose transporter GLUT4 gene expression were assessed in LV tissue. (e) Heart weight and (f) heart weight/body weight. Values are means ± SE. n = 7–21 per group. *p < 0.05; **p < 0.01; ****p < 0.0001 (one-way ANOVA followed by Tukey’s post hoc test).
Effect of insulin treatment on diabetes-induced cardiac oxidative stress
Lucigenin-enhanced, β-NADPH-driven chemiluminescence, a measure of superoxide levels, was increased significantly in diabetic LV (Figure 6(a)). Superoxide levels were reduced to the non-diabetic range in diabetic animals receiving insulin supplementation. Furthermore, p47phox and Nox2 gene expression was also increased in the LV of diabetic rats, both of which were reversed following insulin replacement (Figure 6(b) and (c)).
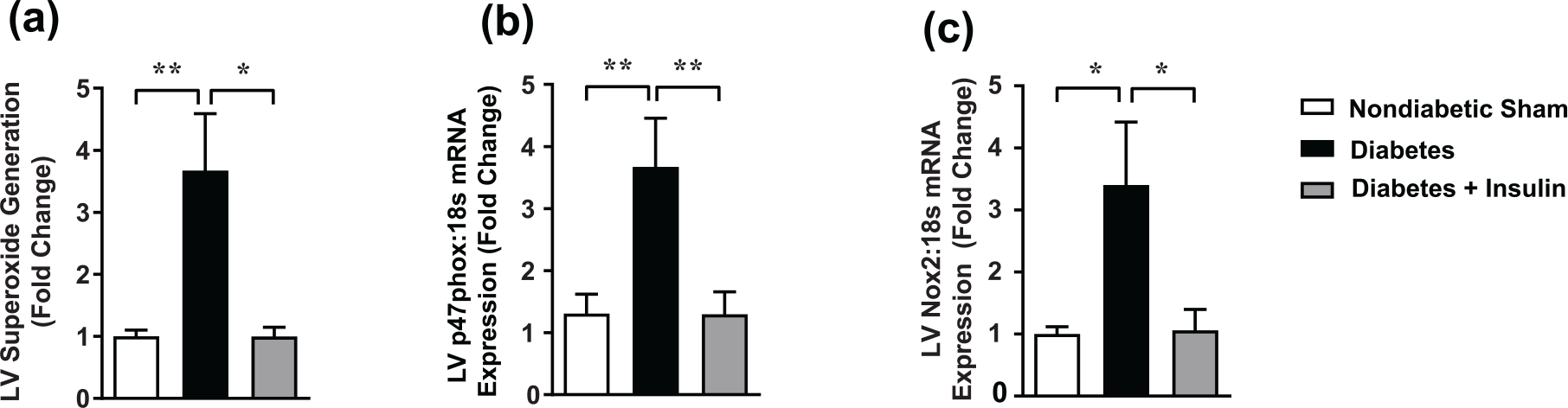
Insulin replacement for the final 4 weeks of 8-week STZ-induced diabetes ameliorates the upregulation of (a) LV NADPH oxidase, including NADPH oxidase activity (LV β-NADPH driven lucigenin-enhanced chemiluminescence superoxide detection), (b) LV p47phox gene expression and (c) LV Nox2 subunit gene expression. Values are means ± SE. n = 8–25 per group. *p < 0.05; **p < 0.01 (one-way ANOVA followed by Tukey’s post hoc test).
Effect of insulin treatment on diabetes-induced cardiac inflammation
Diabetic rats exhibited increased LV mRNA expression of pro-inflammatory cytokines TNF-α, interleukin-6 (IL-6) and MCP-1 compared to non-diabetic rats (Figure 7(a)–(c)). TNF-α, IL-10 and MCP-1 expression were returned to normal levels in diabetic animals treated with insulin. In the same cohort, however, no differences were seen in TRB3 mRNA expression (Figure 7(d)). Furthermore, diabetes caused an increase in mRNA expression of the potent endogenous anti-inflammatory proteins, IL-10 and annexin-A1 (Figure 7(e) and (f)), as well as the FPR2 (Figure 7(g)). Both effects were reversed following insulin replacement. No differences were observed in mRNA expression of FPR1 in the LV (Figure 7(h)).
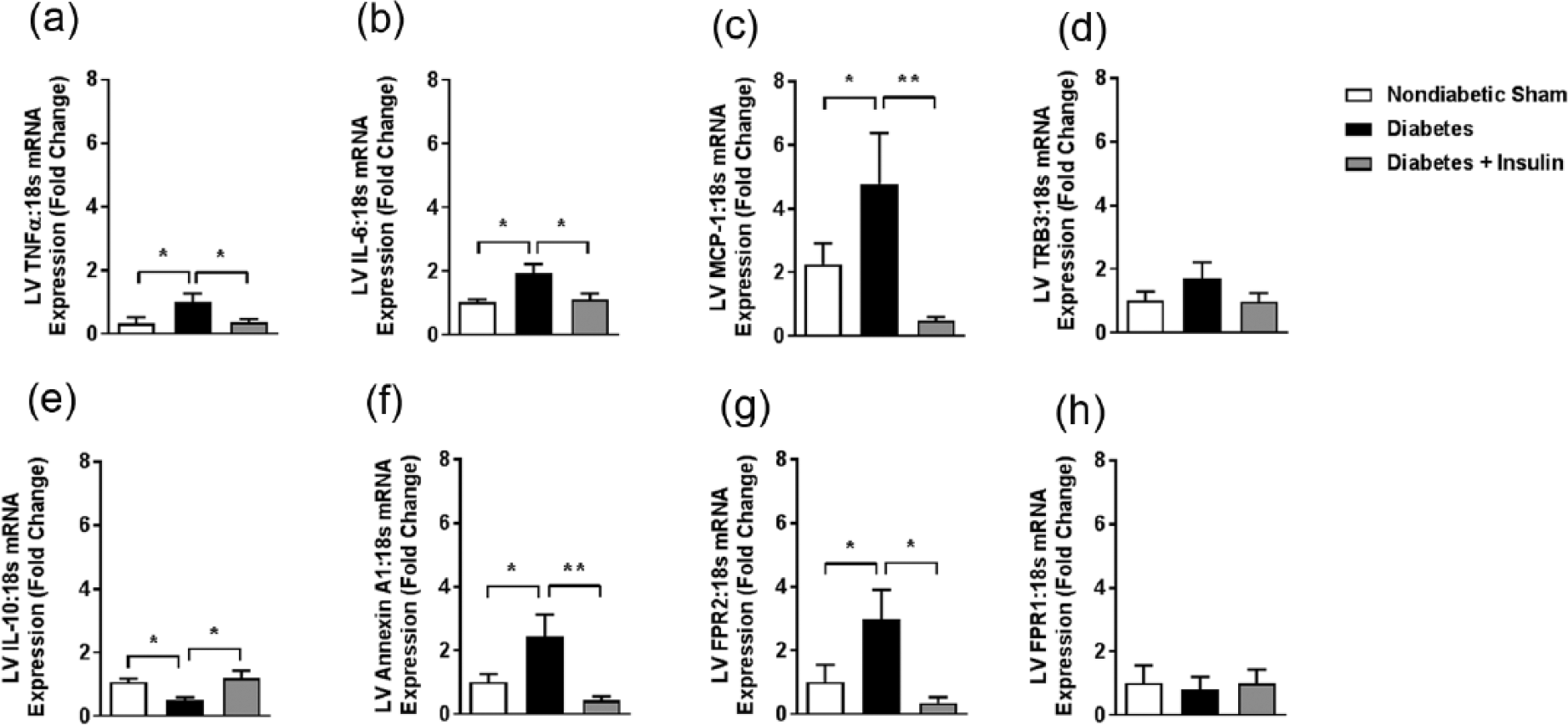
Insulin replacement for the final 4 weeks of 8-week STZ-induced diabetes ameliorates diabetes-induced cardiac inflammation. Gene expression of cardiac inflammatory markers in LV tissue: (a) TNF-α, (b) IL-6, (c) monocyte chemoattractant protein-1 (MCP-1/CCL2), (d) TRB3 and IL-10, (e) Annexin A1, (f) FPR2 and (g) FPR1. Values are means ± SE. n = 8–16 per group. *p < 0.05; **p < 0.01 (one-way ANOVA followed by Tukey’s post hoc test).
Discussion
The most important and clinically relevant finding of this study is the impact of insulin replacement to protect against key cardiac structural and functional changes characteristic of diabetic cardiomyopathy, if given prior to irreversible changes that have limited capacity for repair. Benefits spanned from intracellular redox status, to diastolic functional measures, and histological evidence of myocyte hypertrophy and myocardial fibrosis. This is a timely finding in the context of the increasing global burden of diabetic cardiomyopathy and clinical heart failure, as well as the lack of clarity regarding the pathogenesis of diabetic cardiomyopathy, and the efficacy of pharmacological treatment strategies, targeted against either the underlying metabolic perturbations in diabetes or the heart’s response to stress.
One study described that early type 1 diabetic cardiomyopathy in Akita mice is characterised by diastolic dysfunction and associated with lipotoxicity. 27 However, this animal model has preserved systolic function and is absent of interstitial fibrosis and cardiac hypertrophy. 27 The low-dose STZ rodent model is a particularly attractive model to study the effects of hyperglycaemia on the myocardium without hyperinsulinemia, as it shares common clinical features of diabetic cardiomyopathy, including the presence of interstitial fibrosis and cardiomyocyte hypertrophy. 28 Although type 2 diabetes is arguably more clinically relevant, only the db/db mouse model displays characteristic cardiac dysfunction, but this is complicated by the presence of obesity and impaired leptin signalling. 29 The low-dose STZ protocol circumvents STZ toxicity caused by the high-dose protocol, 30 producing progressive β-cell damage, local inflammation and insulitis, similar to clinical diabetes.
Under normal physiological conditions, insulin in the heart regulates glucose transport, glycolysis, lipid metabolism, glycogen and protein synthesis, myocardial growth, contractility and apoptosis. 31 In this study, diabetic rats displayed an acute increase in plasma glucose levels (Table 1) and observable changes in behaviour including polyphagia, polydipsia and polyuria (Figure 1). Importantly, exogenous insulin introduction significantly improved the diabetic phenotype and had a beneficial effect on plasma glucose levels and urine levels (Figure 1).
The early stages of diabetic cardiomyopathy are characterised by LV diastolic dysfunction and ventricular hypertrophy, and in later stages by LV systolic dysfunction progressing to decompensated heart failure.13,32 The chronic metabolic disturbances caused by diabetes are initially met by short-term physiological adaptation; however, if these persist, the heart undergoes degenerative changes which have limited capacity for repair. In isolated LV papillary muscles from high-dose STZ-induced rats, 10 days and 28 days of insulin therapy was able to partially, and completely, reverse mechanical performance, respectively. 33 Utilising both echocardiography and catheterisation, this study observed significant diastolic dysfunction at the 8-week time point after diabetes induction. Insulin treatment, commencing 4 weeks prior to endpoint, was able to significantly attenuate diabetes-induced reductions in E/A ratio and LV-dP/dt (Figure 2). Furthermore, diabetic animals displayed prolonged aortic ejection time, as well as reductions in LV + dP/dt and LVSP, effects similarly reversed by insulin treatment (Figure 3). These findings are consistent with a previous study which suggested that insulin treatment in STZ diabetic rats prevented their progression to LV systolic dysfunction. 34 Importantly, we now demonstrate that insulin supplementation also preserves LV diastolic function in STZ-induced diabetic rats, on both Doppler flow echocardiography and cardiac catheterisation, back to levels comparable to those seen in rats without diabetes. Having said this, there are conflicting reports on the presence of LV systolic dysfunction in the STZ low-dose diabetes protocol in mice.24,32,35 These differences may be due to species variability or be temporal in nature. Despite this, LV systolic dysfunction is known to succeed diastolic dysfunction at later time points in the STZ-induced mouse model, and in human diabetic cardiomyopathy patients. 32 Impaired cardiac relaxation resulting from LV diastolic dysfunction is often the earliest observable manifestation in human diabetic cardiomyopathy patients. 13 Conventional diagnostic m-mode echocardiography does not adequately detect early LV diastolic dysfunction; hence, this is often undiagnosed in affected patients, unless more sensitive detection techniques are used (e.g. strain, strain rate and myocardial tissue velocity echocardiography).12,31 This difficulty in recognising diabetic cardiomyopathy patients early in its progression, and specifically before other, often more acute forms of cardiovascular disease manifest, 36 has limited our understanding of the distinct pathogenesis of diabetic cardiomyopathy.
Two of the defining and most prominent features of diabetic cardiomyopathy are LV fibrosis and cardiomyocyte hypertrophy.37,38 Insulin regulates the passive mechanical properties of cardiomyocytes in the development of diabetic cardiomyopathy, with absent or dysregulated signalling likely contributing to the remodelling process. In this study, cardiomyocyte hypertrophy was present in the diabetic animals, as measured by cardiomyocyte width (Figure 4), heart weight (normalised to body weight), and by the expression of pro-hypertrophy markers ANP, β-myosin heavy chain and BNP (Figure 5). Interestingly, insulin treatment effectively reversed these adverse effects. Furthermore, insulin treatment exerted an anti-fibrotic effect, with LV collagen deposition reduced in insulin-treated diabetic animals compared to diabetic animals (Figure 4).
The cellular and molecular mechanisms underlying diabetic cardiomyopathy have yet to be fully elucidated, although several putative mechanisms have been proposed, including metabolic disturbances, myocardial fibrosis, small vessel disease and autonomic dysfunction, as well as insulin resistance. Insulin deficiency caused by STZ-induced β-cell destruction prevents insulin-mediated glucose uptake and triggers a compensatory switch to fatty acid oxidation, ultimately leading to impaired contractile function. 39 Furthermore, insulin signalling via the phosphoinositide 3-kinase (PI3K)-α/AKT pathways is important for the translocation of GLUT4 to the plasma membrane. In this study, diabetes caused a decrease in LV GLUT4 expression that was reversed, at least in part, with insulin treatment (Figure 5). However, whether the reversal in GLUT4 expression was directly due to insulin signalling or secondary to improvements in LV contractility requires further investigation.
As alluded to above, a major role has been ascribed to the overproduction of reactive oxygen species (ROS) derived from NADPH oxidase in diabetic cardiomyopathy. 40 In fact, our group has recently demonstrated that LV PI3Kα protects the heart against diabetic cardiomyopathy by specifically reducing LV NADPH oxidase expression and activity. Furthermore, we showed that transgenic mice with reduced cardiac PI3Kα signalling exhibited exaggerated diabetic cardiomyopathy and increased NADPH oxidase function. 41 Similarly, this study describes insulin-mediated reductions in LV superoxide production (Figure 6). Furthermore, insulin treatment attenuated increases in p47phox and Nox2 expression levels present in diabetic rats (Figure 6). Inflammation, and particularly the release of cytokines including TNF-α and IL-1β, has also been implicated in the development of diabetic cardiomyopathy.42,43 In fact, NADPH oxidase-derived ROS have the ability to activate redox-sensitive transcription factors, primarily NF-κβ, causing the release of these pro-inflammatory cytokines. 44 Here, we found the pro-inflammatory cytokines TNF-α and IL-6 to be increased in diabetic rats but reversed by insulin treatment (Figure 7).
Current treatment strategies for heart failure patients with and without diabetes are the same, despite the underlying aetiology being distinct. With recent studies establishing that certain aspects of the pathogenesis of diabetic cardiomyopathy are unique, 45 this suggests that strategies tailored towards diabetic patients may yield more beneficial cardiovascular outcomes. This is the first study to comprehensively characterise insulin replacement on the development of clinically relevant features of diabetic cardiomyopathy in the low-dose STZ-induced diabetic rat. Furthermore, despite the results of this study suggesting that many of the aberrations caused by STZ-induced diabetes in this model are improved by insulin treatment, large-scale clinical studies have failed to show an improvement in major cardiovascular events in diabetic patients following intensive glucose control in diabetes patients.20,46 Overall, since insulin replacement commenced at a relatively early time point in our experimental model, this implies that identifying diastolic dysfunction in patients earlier, using more advanced screening techniques, may provide a therapeutic window that intensive glucose-lowering strategies or pharmacological approaches may prove more effective.
Footnotes
Acknowledgements
The authors acknowledge the Monash Micro-Imaging (MMI) facility for provision of instrumentation and training.
Author contribution
R.H.R. contributed to conception and design of research; M.T., A.H.C., S.G.H., H.K., K.H., M.D., T.L.J. and R.H.R. performed experiments; M.T. and R.H.R. analysed data, prepared figures and drafted manuscript; M.T., X.-J.D. and R.H.R. interpreted results of experiments; M.T., A.H.C., S.G.H., H.K., K.H., M.D., T.L.J., X.-J.D., G.A.F., G.J.D., D.M.K. and R.H.R. approved final version of manuscript; M.T., H.K., X.J.D., G.A.F., G.J.D., D.M.K. and R.H.R. edited and revised manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported in part by a grant-in-aid from Diabetes Australia and by the Victorian Government of Australia’s Operational Infrastructure Support Program. R.H.R. (ID1059960) was supported by Senior Research Fellowship from the National Health and Medical Research Council (NHMRC) of Australia (ID1059660). G.A.F. was co-funded by a Career Development Fellowship from the NHMRC, and a Heart Foundation (of Australia) Future Leader Fellowship.