
Abstract
Heart failure is now recognized as a progressive disease in which patients transition through the stages of being at risk of heart failure (stage A), to asymptomatic structural heart disease (stage B), to clinical manifestations of heart failure (stage C) and finally end-stage or refractory heart failure (stage D). This review outlines the key role of diabetes mellitus as a stage A risk factor for heart failure with preserved ejection fraction, and asymptomatic diabetic cardiomyopathy, referring to the presence of left ventricular diastolic dysfunction in diabetic patients without coronary artery disease, hypertension or other potential aetiologies, as an expression of stage B heart failure with preserved ejection fraction at high risk of transitioning to symptomatic stage C heart failure with preserved ejection fraction. The data presented call for better recognition of the unique phenotype of diabetic cardiomyopathy with preserved ejection fraction and elevated diastolic stiffness as a manifestation of stage B heart failure with preserved ejection fraction that should be targeted for risk management and preventive strategies.
Introduction
Beginning in 2001, the American College of Cardiology and the American Heart Association introduced a new staging system for heart failure (HF). 1 Analogous to the staging system of cancer, this system recognizes HF as a progressive disease in which patients transition through the stages of (1) being at high risk of the development of HF (stage A), (2) developing structural heart disease but without signs or symptoms of HF (stage B), (3) manifesting clinical symptoms of HF (stage C) and finally (4) progressing to end-stage or refractory HF (stage D) (Figure 1). Importantly, the HF stages emphasize that there are established risk factors and structural prerequisites for the development of HF and that therapeutic interventions performed even before the appearance of left ventricular (LV) dysfunction or symptoms can reduce the morbidity and mortality of HF. The staging system therefore serves as a reminder to physicians of the importance of early identification of patients at risk of the development of HF, with the ultimate aim of preventing progression to higher stages of disease.
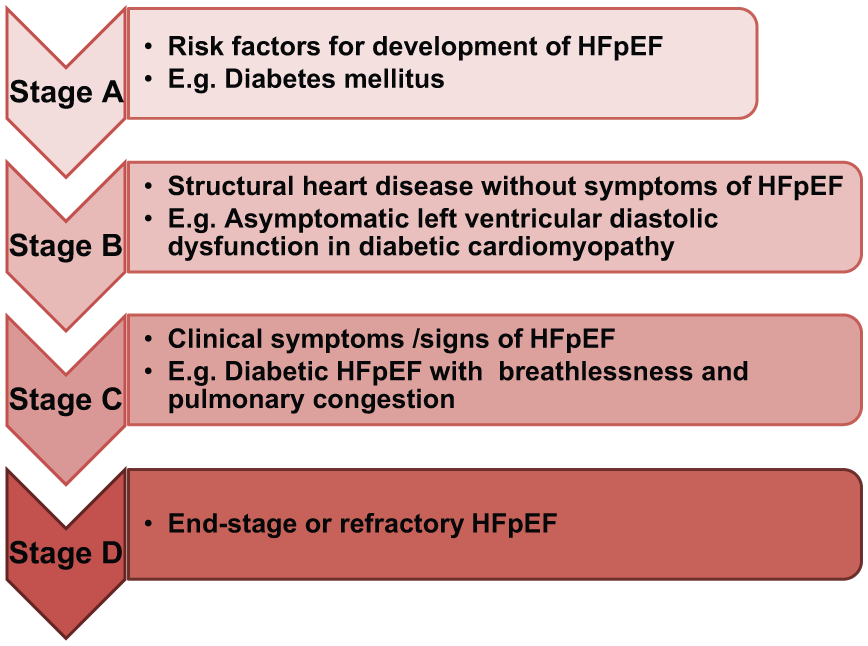
Stages of heart failure. Stages of heart failure (HF) as applied to HF with preserved ejection fraction (HFpEF) and using diabetes mellitus as an example.
Classically, the HF staging system as applied to HF with reduced ejection fraction (HFrEF or ‘systolic HF’) would include patients with coronary artery disease in stage A, patients with a previous myocardial infarction and asymptomatic LV systolic dysfunction in stage B, patients with symptoms and signs of HF in association with LV systolic dysfunction [ejection fraction (EF) < 40%] in stage C and end-stage ischaemic dilated cardiomyopathy requiring specialized treatment strategies such as mechanical circulatory support, continuous inotropic infusions, cardiac transplantation or hospice care in stage D. However, it is now known that half of patients with HF have a preserved EF (EF ⩾ 50%) (HFpEF), with the proportion of HFpEF relative to HFrEF increasing over time, especially in ageing societies where it is projected to become the predominant form of HF. 2
Within this framework, this review seeks to outline the key role of diabetes mellitus (DM) as a stage A risk factor for HF with preserved EF (HFpEF), and asymptomatic diabetic cardiomyopathy, referring to the presence of LV diastolic dysfunction in diabetic patients without coronary artery disease, hypertension or other potential aetiologies, as a manifestation of stage B HFpEF at high risk of transitioning to symptomatic stage C HFpEF.
Epidemiology of DM and HFpEF
DM is projected to reach pandemic proportions over the next few decades, with a World Health Organization (WHO)–estimated global prevalence of 330 million in 2025. 3 Among elderly patients with DM, there is an alarmingly high prevalence, incidence and mortality of HF: HF was prevalent in 22.3% of 151,738 diabetic Medicare beneficiaries aged ⩾65 years in 1994, with an incidence rate of HF of 12.6 per 100 person-years and a mortality rate of 32.7 per 100 person-years (compared with a mortality rate of only 3.7 per 100 person-years among those with diabetes who remained HF-free). 4 Among patients with HFpEF, DM commonly coexists with hypertension, obesity and older age, with a prevalence averaging one-third of HFpEF patients across epidemiologic studies in Western populations 5 and reaching 46% in the large prospective multicentre New York HF registry cohort inclusive of a significant proportion of black non-Hispanic patients. 6
Pathophysiology of HFpEF: HFpEF as an inflammatory cardiometabolic disease
The traditional neuroendocrine model of HF, typified in HFrEF, has been updated in a novel paradigm for HFpEF in which HFpEF is viewed as an inflammatory cardiometabolic disease. 7 According to this paradigm, (1) comorbidities such as DM, obesity and hypertension induce a systemic proinflammatory state with vascular inflammation and endothelial dysfunction; (2) endothelial dysfunction involving the coronary vasculature and central cardiac endothelium limits nitric oxide (NO) bioavailability to adjacent cardiomyocytes; and (3) limited NO bioavailability decreases cyclic guanosine monophosphate (cGMP) production and protein kinase G (PKG) activity in cardiomyocytes, leading to the undesirable effects of LV hypertrophy and diastolic stiffening seen in HFpEF (Figure 2).7,8
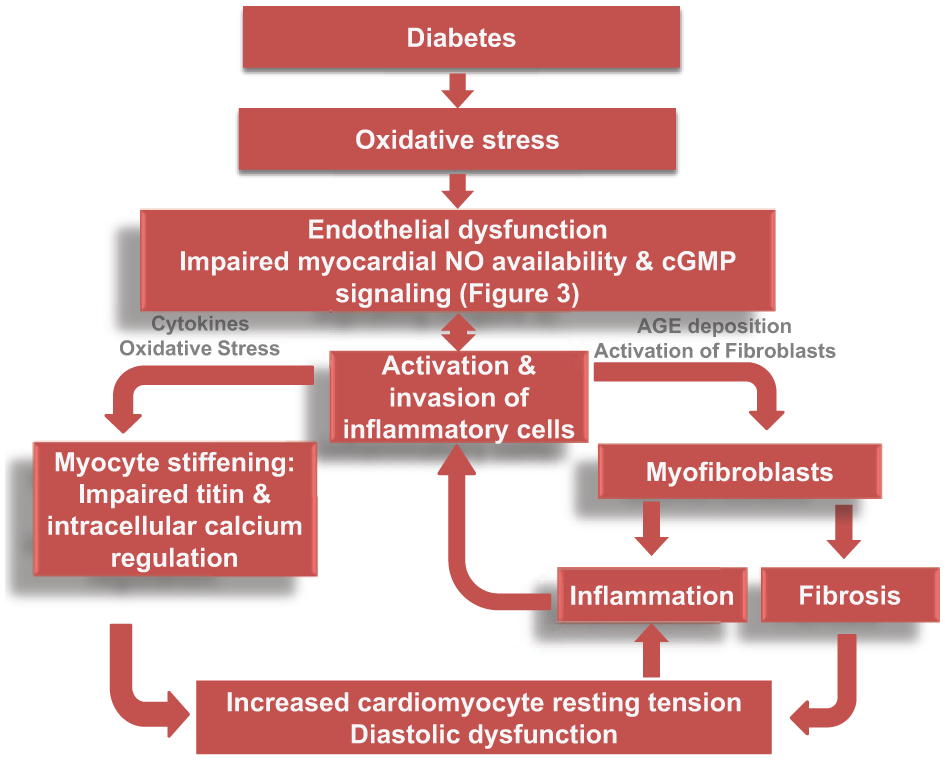
Key role of diabetes in the pathophysiology of HFpEF.
Key role of DM in the pathophysiology of HFpEF
DM induces oxidative stress, vascular inflammation and endothelial dysfunction
In DM, increased oxidative stress occurs via several mechanisms including formation of advanced glycation end products, glucose auto-oxidation, increased levels of free fatty acid and leptin and activation of the polyol pathway. 9 Cardiovascular sources of reactive oxygen species (ROS) production include xanthine oxidoreductase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, mitochondrial oxidases and uncoupled NO synthase. 10 Increased production of ROS (e.g. superoxide, hydrogen peroxide and the hydroxyl radical), coupled with impaired antioxidant defence mechanisms in DM, results in vascular inflammation as evidenced in the enhanced expression of interleukin-6, vascular cellular adhesion molecule-1 and monocyte chemoattractant protein in DM. 11
Under states of oxidative stress, superoxide directly inactivates NO, producing peroxynitrite which contributes to cardiovascular complications of DM and HF. 12 Furthermore, peroxynitrite oxidizes tetrahydrobiopterin – a necessary cofactor regulating the function of endothelial NO synthase. Reduced tetrahydrobiopterin results in endothelial nitric oxide synthase (eNOS) uncoupling, causing a vicious cycle where more superoxide is produced instead of NO, thus amplifying oxidative stress and endothelial dysfunction. 13
Central cardiac endothelial dysfunction limits NO bioavailability to adjacent cardiomyocytes
In HFpEF, endothelial dysfunction is highly prevalent, correlates with functional status and predicts cardiovascular events in HFpEF.14,15 Recently, the vascular endothelial product NT-pro C-type natriuretic peptide was shown to be strongly predictive of outcomes in HFpEF but not HFrEF, further supporting a pathophysiologic role of endothelial dysfunction in HFpEF. 16
Importantly, the consideration of the role of endothelial dysfunction in HFpEF should go beyond that of the peripheral endothelium (endothelial cells in various peripheral organs) and include careful consideration of the central cardiac endothelium (endothelial cells of the coronary vessels, intramyocardial capillaries and intracardiac endocardium). 17 In fact, the cardiac endothelium, along with the pulmonary vascular endothelium, is the largest endothelial surface of the body, and both cardiac endothelial dysfunction and pulmonary vascular endothelial dysfunction have been shown to contribute to the development of HF. 17
Cardiac (endocardial) endothelial dysfunction results in reduced NO bioavailability to the adjacent cardiomyocytes, leading to reduced NO-mediated activation of soluble guanylate cyclase which generates cGMP (Figure 3). cGMP is an important second messenger that modulates cardiac structure and function via activation of its downstream effectors, including PKG. PKG activation results in attenuation of myocardial hypertrophy, decreased myofilament calcium sensitivity, pro-lusitropy and anti-inflammatory effects. Indeed, both low cGMP and low PKG activity have been demonstrated in myocardial biopsies of patients with HFpEF, compared to that of HFrEF or aortic stenosis (pure pressure overload). 18 Furthermore, lower myocardial PKG correlated with larger cardiomyocyte diameter in HFpEF compared to HFrEF, 18 and increasing myocardial cGMP, via inhibition of its breakdown by phosphodiesterase-5, reversed cardiomyocyte hypertrophy in a mouse model of HFpEF. 19 The latter highlights the importance of the myocardial NO-cGMP-PKG pathway as a potential therapeutic target, as evidenced by the current therapeutic strategies being tested in clinical trials of HFpEF (Figure 3). 20
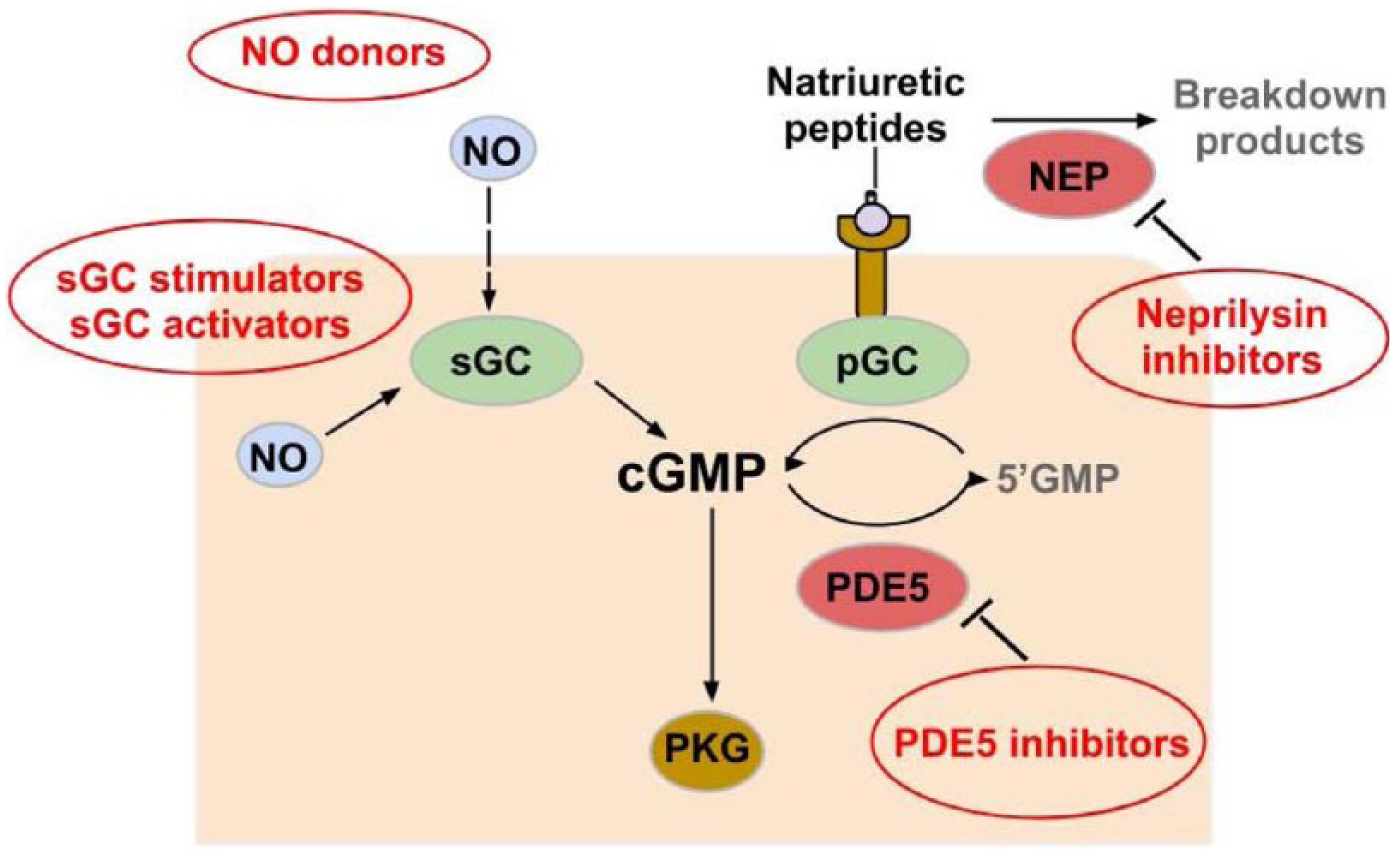
Role of nitric oxide (NO) – cyclic guanosine 3′,5′-monophosphate (cGMP) – protein kinase G (PKG) activity pathway in HFpEF.
Diabetic cardiomyopathy as stage B HFpEF
Evidence from experimental studies in isolated cardiomyocytes
The key role of DM in HFpEF was investigated in an elegant study by Van Heerebeek et al., 21 where endomyocardial biopsy samples were compared between 28 patients with HFpEF (16 with DM) and 36 patients with HFrEF (10 with DM), all without coronary artery disease. Compared to non-diabetic patients, diabetic HF patients had higher LV diastolic stiffness irrespective of EF. In diabetic HFpEF, increased LV diastolic stiffness was predominantly due to increased cardiomyocyte resting tension of hypertrophied cardiomyocytes; whereas cardiomyocyte resting tension was similar in diabetic and non-diabetic HFrEF. Of note, cardiomyocyte hypertrophy in the diabetic HFpEF patients was not attributable to increased LV pressure overload, and DM was the specific cause of increased LV diastolic stiffness in the subgroup of diabetic HFpEF patients who did not have arterial hypertension. This subgroup clearly exemplified the unique phenotype of diabetic cardiomyopathy with preserved LV EF and elevated diastolic LV stiffness without LV dilatation, that is, the expected phenotype in stage B of HFpEF. Thus, diabetic cardiomyopathy should not merely be regarded as a condition of LV dilatation and reduced EF (‘dilated cardiomyopathy’); instead, its definition should importantly include LV diastolic dysfunction (with preserved EF) as a prominent manifestation. 22
Van Heerebeek et al. 21 further demonstrated that the increased cardiomyocyte resting tension in diabetic HFpEF was corrected by protein kinase A (PKA), indicating that the high resting tension was due to a phosphorylation deficit of the myofilamentary or cytoskeletal protein of the cardiomyocyte which could therefore be reversed by administration of PKA.21,23 An important target of PKA phosphorylation is titin – the giant sarcomeric elastic protein spanning from the Z-disc to the M-line of the cardiomyocyte and serving as a stretch/stress sensor that transmits external forces from the extracellular matrix to the cardiomyocyte skeleton and determines cardiomyocyte resting tension. 24 Phosphorylation of the N2B region of titin by PKA has been shown to reduce myofibrillar resting tension in cardiomyocytes isolated from both human and experimental HFpEF. 24 Supporting the key role of titin phosphorylation changes in HFpEF, increased expression and lower phosphorylation of the stiff N2B titin isoform have been demonstrated in cardiomyocytes isolated from patients with HFpEF. 23 Further supporting titin’s role in diabetic HFpEF specifically, resting tension in cardiomyocytes from diabetic HFpEF patients was shown to correlate with opening of the cardiomyocyte Z-discs, which were wider in diabetic compared to non-diabetic HFpEF, 21 indicating altered elastic properties of the cytoskeletal proteins which pull at the ends of the Z-discs. 24
Similar to PKA administration, administration of PKG to cardiomyocytes isolated from patients with HFpEF has also been shown to reduce resting tension.18,25 In fact, there was no further reduction in cardiomyocyte resting tension when PKA was administered after PKG, suggesting that PKG and PKA act on the same phosphorylation sites. 25 In aggregate with the evidence described above of prominent oxidative stress, vascular inflammation, endothelial dysfunction and reduced NO bioavailability to cardiomyocytes in DM, the NO-cGMP-PKG-titin pathway is likely to be a major determinant of LV diastolic stiffness in diabetic HFpEF patients.
Evidence from clinical studies in epidemiologic and clinical trial patient cohorts
Epidemiological evidence of longitudinal progression from stage B to stage C of HFpEF was provided in 1038 participants of the Framingham Heart Study original cohort, in whom antecedent LV diastolic dysfunction was independently related to future incident HFpEF over an average 11 years of follow-up. 26 Specifically in DM, a large community-based cohort of 1760 diabetic patients in Olmsted County, MN, all studied using Doppler echocardiography, was followed for incident HF. 27 A strikingly high prevalence of asymptomatic LV diastolic dysfunction (i.e. stage B HFpEF, present in 23%) was found among these community-based diabetic adults. Increasing severity of LV diastolic dysfunction was independently related to increasing risk of subsequent incident HF, with the cumulative probability of the developing HF within 5 years of 36.9% versus 16.8% in diabetic patients with versus without diastolic dysfunction (p<0.001). Additionally, diabetic patients with diastolic dysfunction had a significantly higher mortality compared to those without diastolic dysfunction. 27 Further evidence of the role of DM in the progression to adverse outcomes in stage C HFpEF was recently provided in a sub-study of the RELAX (Phosphodiesterase-5 inhibition to improve clinical status and exercise capacity in HF with preserved EF) study. 28 Compared to non-diabetic HFpEF patients, diabetic HFpEF patients had reduced exercise capacity and increased risk of hospitalization, associated with a more severe disease phenotype characterized by greater LV hypertrophy, and elevated circulating markers of oxidative stress, inflammation and fibrosis. Notably, the association of DM with LV diastolic dysfunction is not limited to established or advanced DM, but also exists along the entire spectrum of glucose metabolism from pre-diabetic to non-insulin-treated and insulin-treated DM. 29 These findings have important implications for preventive approaches, which are especially critical in HFpEF since there is, to date, still no proven effective treatment for stage C HFpEF once it is established.
Conclusion
The data presented in this review call for healthcare providers involved in the active management of diabetic patients, including generalists, internists, endocrinologists and cardiologists, to recognize the unique phenotype of diabetic cardiomyopathy with preserved LV EF and elevated diastolic LV stiffness without LV dilatation, that is, diabetic cardiomyopathy as a manifestation of stage B of HFpEF. This differs from prior definitions where diabetic cardiomyopathy was regarded as a condition of LV dilatation and reduced EF (‘dilated cardiomyopathy’). Importantly, patients with diabetic stage B HFpEF are at risk of further progression to symptomatic stage C HFpEF. Awareness and identification of patients at risk are the first steps towards the ultimate goal of optimal management to prevent or delay progression of HF in patients with DM.
Footnotes
Declaration of conflicting interests
The authors declare that they have no conflicts of interest.
Funding
C.S.P.L. is supported by a Clinician Scientist Award from the National Medical Research Council of Singapore; has received research support from Boston Scientific, Medtronic and Vifor Pharma and has consulted for Novartis, Bayer, Astra Zeneca and Vifor Pharma.