
Abstract
Hydrogen sulphide (H2S) is endogenously produced in vascular tissue and has anti-oxidant and vasoprotective properties. This study investigates whether chronic treatment using the fast H2S donor NaHS could elicit a vasoprotective effect in diabetes. Diabetes was induced in male C57BL6/J mice with streptozotocin (60 mg/kg daily, ip for 2 weeks) and confirmed by elevated blood glucose and glycated haemoglobin levels. Diabetic mice were then treated with NaHS (100 µmol/kg/day) for 4 weeks, and aortae collected for functional and biochemical analyses. In the diabetic group, both endothelium-dependent vasorelaxation and basal nitric oxide (NO•) bioactivity were significantly reduced (p < 0.05), and maximal vasorelaxation to the NO• donor sodium nitroprusside was impaired (p < 0.05) in aorta compared to control mice. Vascular superoxide generation via nicotine adenine dinucleotide phosphate (NADPH) oxidase (p < 0.05) was elevated in aorta from diabetic mice which was associated with increased expression of NOX2 (p < 0.05). NaHS treatment of diabetic mice restored endothelial function and exogenous NO• efficacy back to control levels. NaHS treatment also reduced the diabetes-induced increase in NADPH oxidase activity, but did not affect NOX2 protein expression. These data show that chronic NaHS treatment reverses diabetes-induced vascular dysfunction by restoring NO• efficacy and reducing superoxide production in the mouse aorta.
Introduction
In mammalian tissues, reactive oxygen species (ROS) are produced in both pathological and physiological conditions. The parent ROS molecule superoxide
Vascular dysfunction, of both micro- and macro-vessels, is a major cause of morbidity in diabetes. Endothelial dysfunction, in both conductance and resistance arteries, is an initiating factor in the pathogenesis of vascular disease in both type 1 and type 2 diabetes7,8 There is considerable evidence that hyperglycaemia leads to oxidative stress resulting in endothelial dysfunction.9,10
Hydrogen sulphide (H2S) is well known as a toxic gas with the distinct smell of rotten eggs; however, there is now an abundance of literature to indicate that H2S is a gasotransmitter with important physiological functions.
11
In particular, there is mounting evidence that H2S is a cytoprotective molecule with anti-inflammatory and anti-oxidant effects,
12
including in the vasculature.
13
Indeed, H2S is a potent one-electron chemical reductant and nucleophile that is reported to be capable of scavenging free radicals.
14
H2S is also important in regulating mitochondrial function15,16 and reduces mitochondrial ROS formation.
17
There is evidence that H2S decreases vascular
The vasoprotective effects of H2S have largely been demonstrated in vitro or in cell-based assays, but data showing that these effects are translated in vivo are lacking. There is evidence of the vasoprotective effects of H2S in hypertension 23 and atherosclerosis, 24 and sodium hydrosulfide (NaHS) treatment can attenuate endothelial dysfunction in diabetic rats. 25 These conditions all involve vascular oxidative stress and the potential protective effects of H2S on the vasculature under oxidative stress need to be further investigated. Therefore, the purpose of this study is to investigate the ability of chronic NaHS treatment to protect vascular function in vivo under conditions of diabetes-induced oxidative stress.
Methods
All experimental procedures were approved by the Animal Experimentation Ethics Committee of RMIT University and conformed to the National Health and Medical Research Council of Australia code of practice for the care and use of animals for scientific purposes.
Induction of diabetes
Male 8-week-old C57BL/6J mice (Animal Resource Centre, Western Australia) were randomly divided into three groups: control, diabetic and diabetic treated with NaHS. Type 1 diabetes was induced with streptozotocin (STZ, 60 mg/kg/day, ip) for 2 weeks, whereas the control group received an equivalent volume of vehicle (0.1 M citrate buffer, pH: 4.5). Five weeks after the commencement of STZ injections, half the diabetic mice were randomly allocated to receive NaHS (100 µmol/kg/day, ip) for a further 4 weeks. Blood glucose level was measured at the beginning and at the end of the experiment and twice a week during the experiment for diabetic mice. Insulin 0.1 U, ip, 50% isophane insulin (Protaphane®) + 50% neutral insulin (Actrapid®) was given when the blood glucose level exceeded 30 mM to prevent excessive loss of weight. Control group blood glucose level was also measured during week 5. Blood samples were obtained from the tail vein and non-fasting blood glucose concentration was measured using a one touch glucometer ACCU-CHEK Advantage® (Roche, NSW, Australia) and glycated haemoglobin (HbA1C) using an in2it (II) analyser (281-0000EX) (Bio-Rad, NSW, Australia). Induction of diabetes was considered successful when the blood glucose level was >25 mM.
Tissue collection
Mice were culled humanely using CO2 asphyxiation (95% CO2, 5% O2) followed by cervical dislocation and decapitation, at which point blood samples were collected for determination of HbA1C and plasma total sulphide concentrations. The whole aorta was isolated and immediately placed in ice cold Krebs bicarbonate solution [composition (mM): NaCl 118, KCl 4.7, MgSO4 1.18, KH2PO4 1.2, NaHCO3 25,
Vascular reactivity studies
Aortic rings of 2-mm long were mounted on a multi-wire myograph system 610 M (Danish Myo Technology, Aarhus, Denmark) filled with Krebs bicarbonate solution. The vessels were allowed to stabilise at zero tension for 10 min before the 15 min equilibration period at 5 mN. All experiments were performed at 37°C and continuously bubbled with carbogen (95% O2 and 5% CO2). Changes in tension were recorded using a Powerlab data acquisition system (ADInstruments, Australia).
Assessment of vascular endothelial and smooth muscle cell function
Vascular function was examined as previously described 26 with the following modifications: Aortic rings were maximally contracted with the thromboxane A2 mimetic, U46619 (1 µM) and once the U46619-induced maximum contraction reached a plateau (Fmax), the aortic rings were washed with Krebs solution to regain their basal tension. Following determination of Fmax, the aortic rings were then pre-contracted to ~50% Fmax using titrated concentrations of U46619 (0.1–100 nM) followed by cumulative concentration–response curves to acetylcholine (ACh, 0.1 nM–10 µM) to assess endothelial function. Endothelium-independent relaxation was examined with the nitric oxide donor sodium nitroprusside (SNP, 0.1 nM–3 µM) and the KATP channel opener levcromakalim (LKM, 0.1 nM–10 µM). Maximal relaxation was achieved by adding the Ca2+ channel blocker nifedipine (10 µM) at the end of each concentration–response curve.
Assessment of basal NO• bioactivity
In a separate set of experiments, the effect of diabetes on basal NO• bioactivity was examined by measuring the contractile response of endothelium intact aortic rings to the NO• synthase inhibitor Nω-Nitro-
Vascular superoxide production
NADPH oxidase–derived superoxide production in the aorta was measured using lucigenin-enhanced chemiluminescence assay. Aortic rings (2 mm) were pre-incubated for 45 min at 37°C in Krebs-HEPES buffer [composition (mM): NaCl 99.9, KCl 4.7, KH2PO4 1.0, MgSO4 1.2,
Western blotting
Western blotting was performed as described previously. 29 Briefly, aortae were excised, snap frozen in liquid nitrogen and homogenised in Laemmli buffer [25% glycerin, 12.5% β-mercaptoethanol, 7.5% sodium docedyl sulphate, 25% 1 mol/L Tris–HCl (pH: 8.0) and 0.25 mg/mL bromophenol blue] over liquid nitrogen. Protein concentration was determined using the reducing agent and detergent compatible (RCDC) assay (Bio-Rad). Equal amounts of protein were loaded onto a 10% polyacrylamide gel and transferred to a nitrocellulose membrane. Membranes were blocked in 5% skim milk for 1 h and then incubated overnight at 4°C with the appropriate primary antibody (1:1000 for eNOS and NOX2, 1:2000 for β-actin) in 5% skim milk. Membranes were then incubated with a horseradish peroxidase–conjugated anti-mouse IgG for 1 h. Immunoreactive bands were detected by enhanced chemiluminescence, quantified using a ChemiDoc XRS molecular imager (Bio-Rad) and normalised to intensity of corresponding bands for β-actin.
Plasma sulphide assay
Mouse plasma samples were added to zinc acetate (1% w/v) followed by the addition of trichloroacetic acid (10% w/v). Subsequently, N,N-dimethyl-p-phenylenediamine sulphate (20 mM) in 7.2 M HCl and FeCl3 (30 mM) in 1.2 M HCl were added to each of the samples. After 20-min incubation at room temperature, the sample solutions were centrifuged at nine relative centrifugal force for 1 min and 250 µL of each sample supernatant was pipetted into a 96-well plate and measured at 670 nm using a spectrophotometer (Thermo Scientific Multiskan Spectrum). The H2S concentration of each sample was calculated against a calibration curve of NaHS (0–200 µM) and results are expressed in micromolar.
Reagents
All drugs were purchased from Sigma-Aldrich (USA), except for acetylcholine perchlorate (ACh, BDH Chemicals, UK), LKM (Tocris, Australia) and U46619 (Sapphire Bioscience, Australia). All drugs were dissolved in distilled water, except LKM and nifedipine which were dissolved in ethanol.
Statistical analyses
All results are expressed as mean ± standard error of mean (SEM), n represents the number of animals per group. Concentration–response curves from mouse aorta experiments were computer fitted to a sigmoidal curve using non-linear regression (Prism version 6; GraphPad Software, USA) to calculate the sensitivity of each agonist (pEC50). Maximum relaxation (Rmax) to ACh, SNP and LKM was measured as a percentage of pre-contraction to U46619, where the response to nifedipine was considered to be 100%. Group pEC50 and Rmax values were compared by one-way analysis of variance (ANOVA) with post hoc analysis using the Holm–Sidak test. p < 0.05 was considered statistically significant.
Results
Body weight and blood glucose levels
Body weight gained, blood glucose level, HbA1C and plasma sulphide levels are shown in Table 1. A total of 9 weeks after commencement of treatment with STZ or citrate buffer, the body weight gain in control mice was significantly greater than in diabetic mice (p < 0.05). The blood glucose and HbA1C levels in diabetic mice were significantly greater than control mice (p < 0.05). Treatment with NaHS had no significant effect on body weight gain, blood glucose or HbA1C levels in diabetic animals. Total plasma sulphide levels at the end of the experiment were not different between the groups.
Body weight, blood glucose and plasma sulphide data.
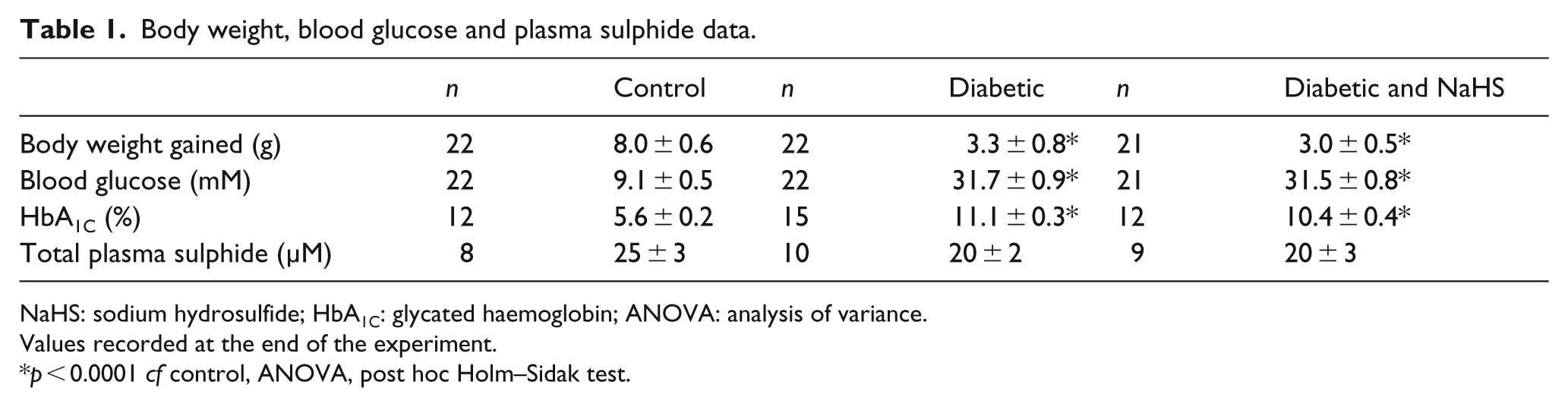
NaHS: sodium hydrosulfide; HbA1C: glycated haemoglobin; ANOVA: analysis of variance.
Values recorded at the end of the experiment.
p < 0.0001 cf control, ANOVA, post hoc Holm–Sidak test.
Effect of diabetes and NaHS treatment on endothelial function
The maximal contraction elicited by U46619 was not affected by diabetes or NaHS treatment (Table 2). Pre-contraction levels for vasorelaxant concentration–response curves were matched between the groups (Table 2). Diabetes significantly reduced the sensitivity and maximal relaxation to ACh (Figure 1(a) Table 2, p < 0.05). Treatment with NaHS in diabetic mice returned the sensitivity and maximal relaxation response to ACh to that of the control group. NO• bioactivity, assayed by the contractile response to L-NAME, was significantly reduced in the diabetic group, but this was also restored with NaHS treatment (Figure 1(b), p < 0.05).
Parameters for vascular reactivity experiments.
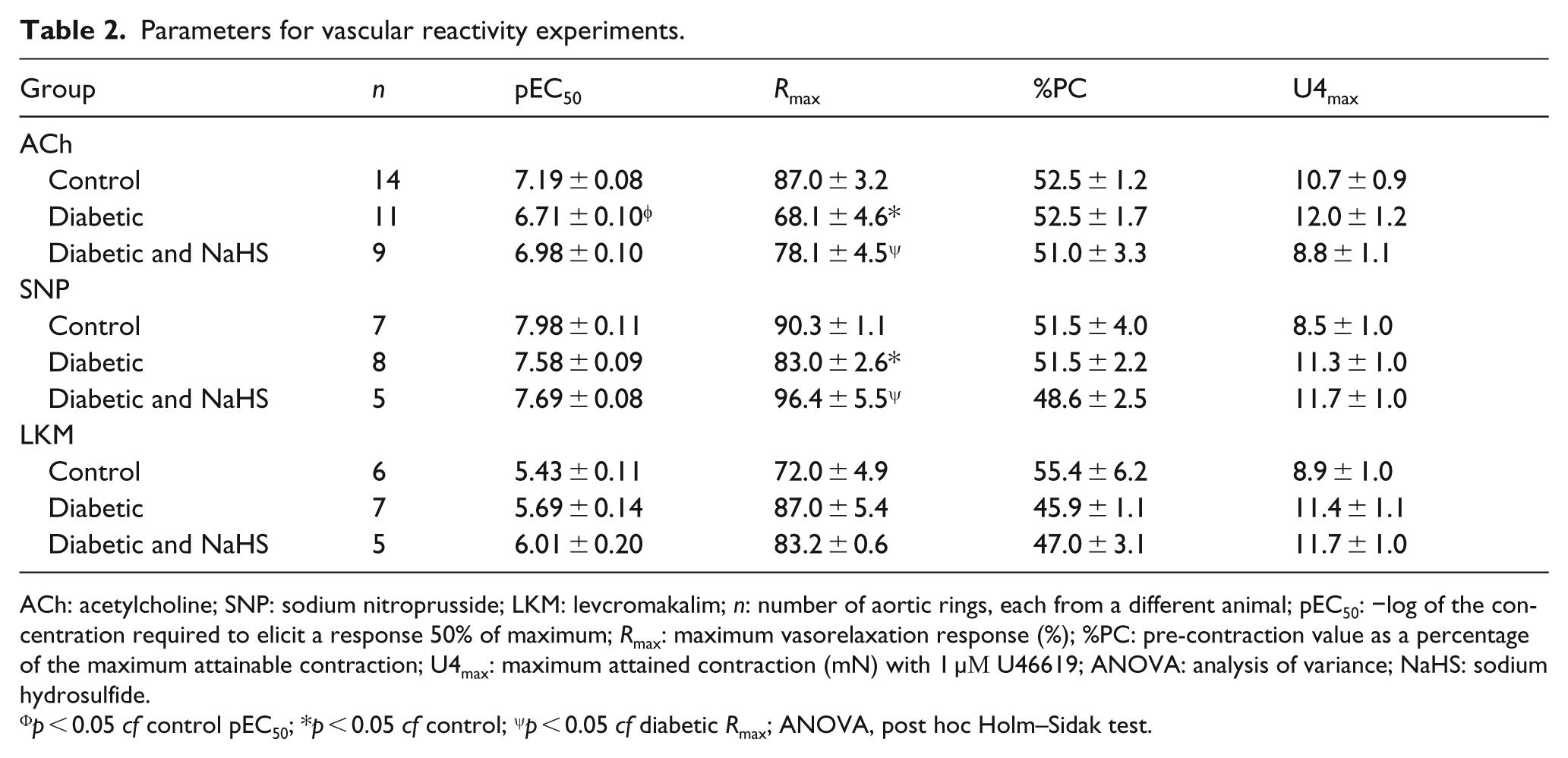
ACh: acetylcholine; SNP: sodium nitroprusside; LKM: levcromakalim; n: number of aortic rings, each from a different animal; pEC50: −log of the concentration required to elicit a response 50% of maximum; Rmax: maximum vasorelaxation response (%); %PC: pre-contraction value as a percentage of the maximum attainable contraction; U4max: maximum attained contraction (mN) with 1 µΜ U46619; ANOVA: analysis of variance; NaHS: sodium hydrosulfide.
p < 0.05 cf control pEC50; *p < 0.05 cf control; ψp < 0.05 cf diabetic Rmax; ANOVA, post hoc Holm–Sidak test.
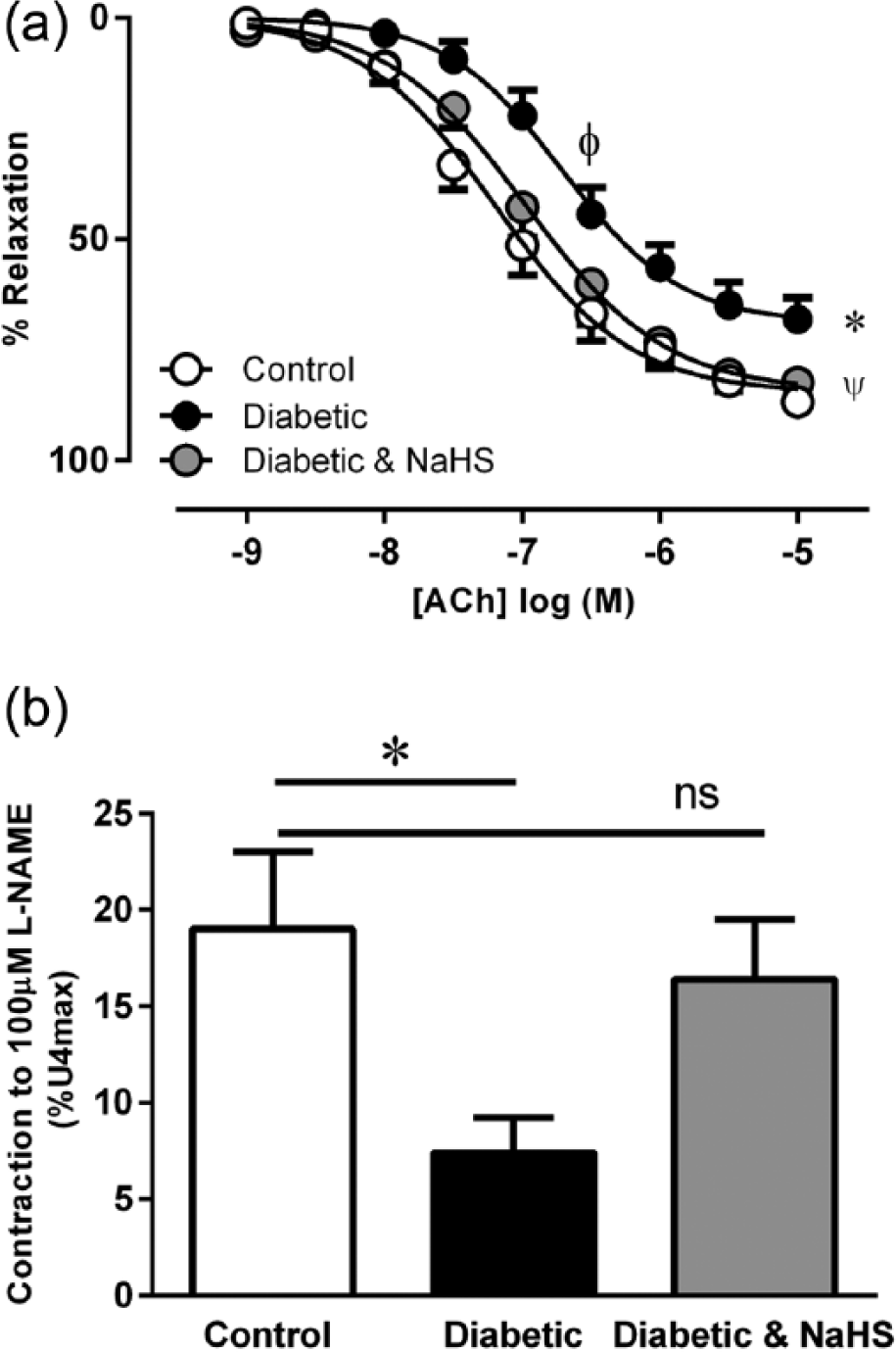
(a) Vasorelaxation response to ACh. φp < 0.05 cf control pEC50, *p < 0.05 cf control Rmax, ψp < 0.05 cf diabetic Rmax, ANOVA, post hoc Holm–Sidak test, n = 9–14. (b) Basal NO• bioactivity in aorta from control, diabetic and diabetic treated with NaHS mice *p < 0.05 cf control, ns = p > 0.05 control cf diabetic and NaHS, ANOVA, post hoc Holm–Sidak test, n = 5–7.
Effect of diabetes and NaHS treatment on vascular smooth muscle cell function
The maximum vasorelaxation response elicited by exogenous NO•, delivered via SNP, was significantly reduced in the diabetic group although the sensitivity was unaffected. The maximum relaxation to SNP was restored in the NaHS treated group (Figure 2(a), Table 2, p < 0.05). The response to the KATP channel opener LKM was neither affected by diabetes nor treatment with NaHS (Table 2).
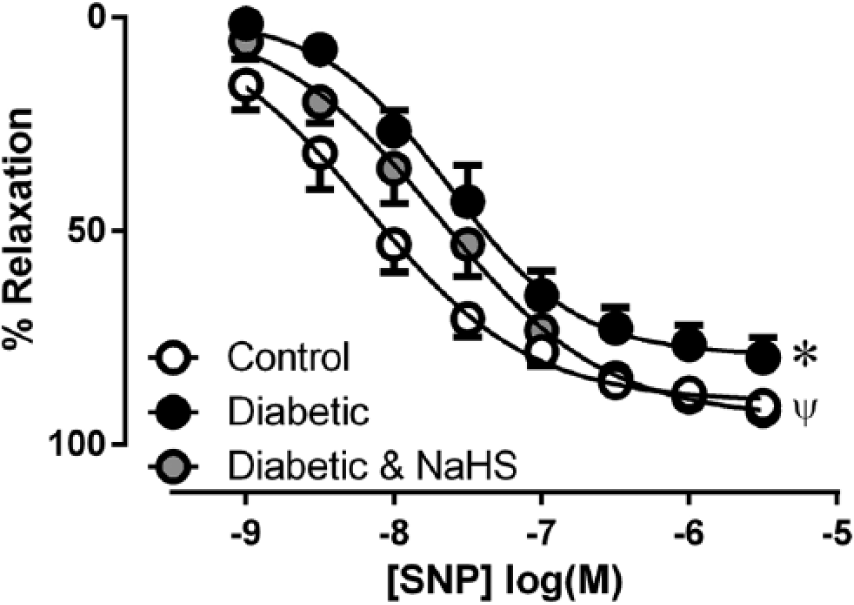
Vasorelaxation response to the NO• donor SNP in aorta from control, diabetic and diabetic treated with NaHS mice. *p < 0.05 cf control Rmax, ψp < 0.05 cf diabetic Rmax, ANOVA, post hoc Holm–Sidak test, n = 5–8.
Effect of diabetes and NaHS treatment on vascular superoxide production
The level of superoxide production detected by lucigenin-enhanced chemiluminescence in aorta from diabetic mice was significantly increased compared to control mice (Figure 3, p < 0.05). NaHS treatment significantly reduced superoxide production from diabetic aorta (p < 0.05). In all groups, superoxide production from aorta could be virtually abolished by DPI, a flavoprotein inhibitor of NADPH oxidase (Figure 3).
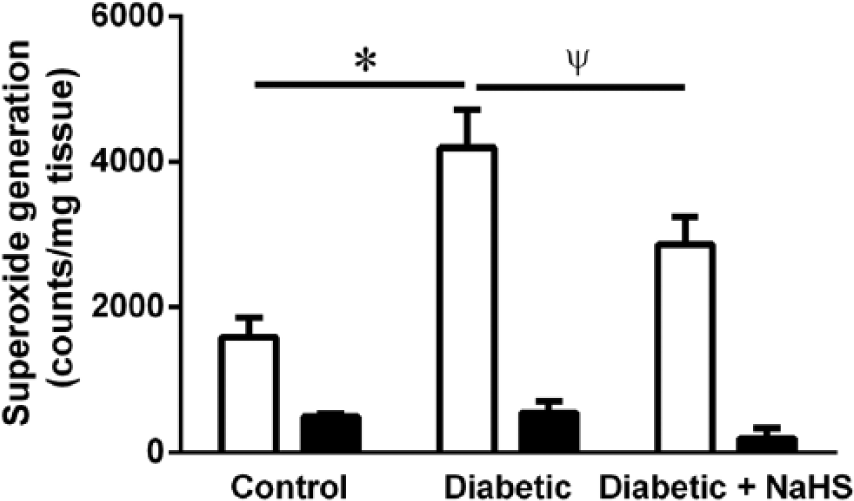
Superoxide production measured by lucigenin-enhanced chemiluminescence in aorta from control, diabetic and diabetic treated with NaHS mice, with (filled bars) and without (open bars) the NADPH oxidase inhibitor DPI. *p < 0.05 cf control, ψp < 0.05 cf diabetic, ANOVA, post hoc Holm–Sidak test, n = 5–8.
Effect of diabetes and NaHS treatment on NOX2 and eNOS protein expression
NOX2 expression was significantly enhanced in the diabetic group, but this was not affected by NaHS treatment (Figure 4, p < 0.05). Total eNOS expression was unaffected by diabetes or NaHS treatment (Figure 4).
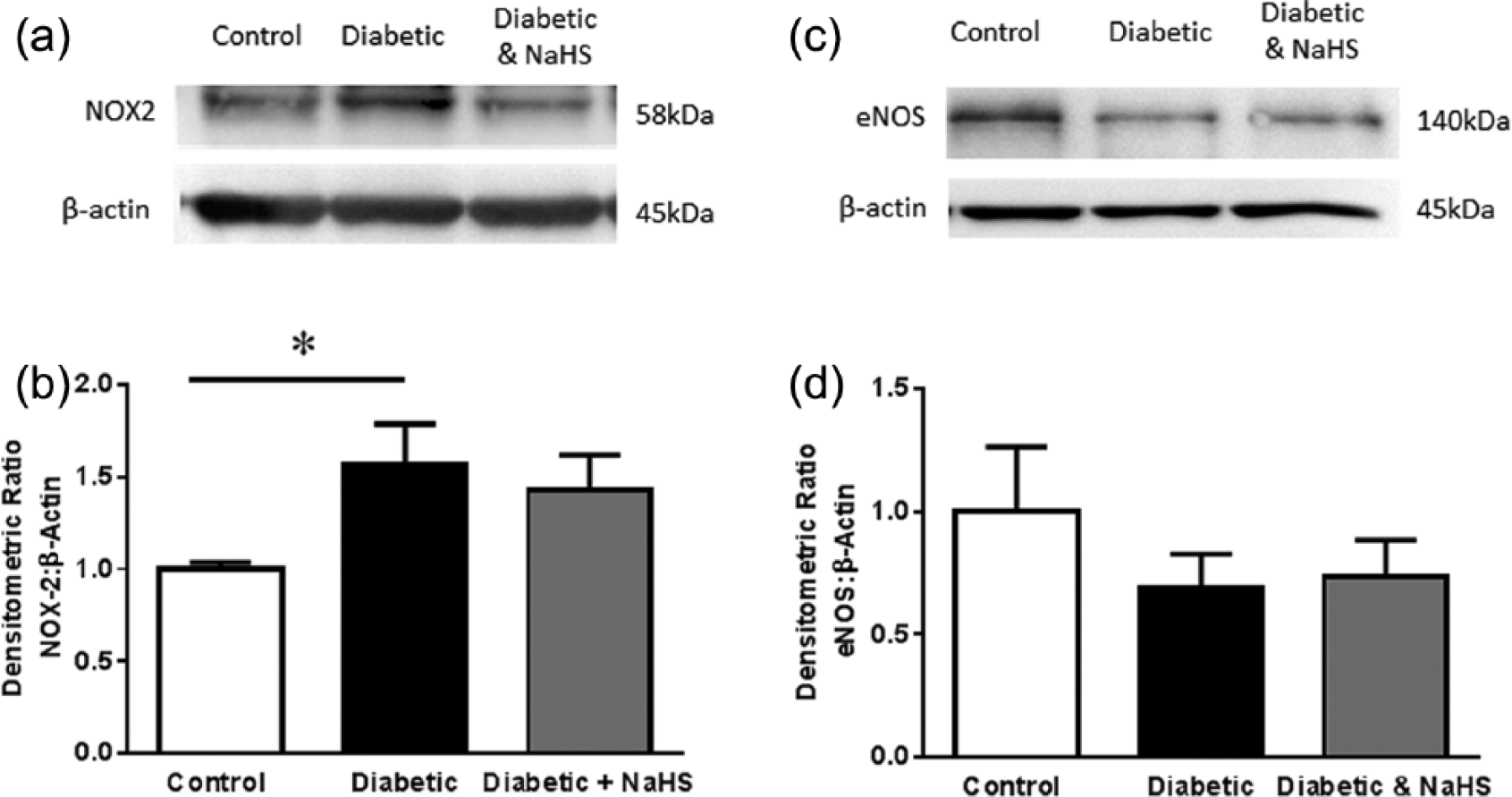
NOX2 expression, normalised to β-actin in aorta from control, diabetic and diabetic treated with NaHS mice. (a) Example western blots from each group. (b). Group data, *p < 0.05 cf control, ANOVA, post hoc Holm–Sidak test, n = 9. eNOS expression, normalised to β-actin in aorta from control, diabetic and diabetic treated with NaHS mice. (c) Example western blots from each group. (d). Group data, p > 0.05, ANOVA, post hoc Holm–Sidak test, n = 9.
Discussion
This study demonstrates the ability of H2S to protect endothelial function under conditions of oxidative stress, in diabetes. Indeed, we have shown that treatment of diabetic animals with the fast H2S donor NaHS reduces vascular superoxide production by inhibiting vascular NADPH oxidase activity. This is associated with an improvement in endothelial function, likely as a result of reduced inactivation of NO• by superoxide. Consistent with this, our data also show that the efficacy of exogenous NO• is protected by NaHS treatment. This is the first study to show the specific protective effects of NaHS on the vasculature in diabetes, where there is increased oxidative stress, and it provides further evidence of the potential role of H2S in regulating vascular NADPH oxidase function in vivo.
Daily NaHS treatment for 4 weeks following induction of diabetes had a marked protective effect on endothelial function, by restoring ACh-mediated vasorelaxation and basal NO• bioactivity, as assessed by the contraction response to L-NAME. This may relate to an effect on eNOS activity or may be due to a protective effect of endogenous NO•, after production. There was no significant difference in total eNOS protein expression with diabetes or NaHS treatment. Previous studies have shown that diabetes reduces the expression of the physiological dimer form of the enzyme 30 which produces NO•, and increases the presence of the monomer form, which is a superoxide generating enzyme 31 further exacerbating oxidative stress. Indeed, H2S has been previously shown to have a stimulatory effect on the NO• signalling pathway since there is in vitro evidence that H2S facilitates the phosphorylation and s-sulfhydration of eNOS 32 as well as increasing the production of NO• from endothelial cells.33,34 This study did not examine the abundance of monomer versus dimer expression of eNOS nor changes in the phosphorylation of the enzyme, which are known to be altered in diabetes 35 and these would be important lines of investigation for future studies.
Consistent with previous reports, we found that diabetes induced vascular oxidative stress as evidenced by increased NADPH-oxidase dependent vascular superoxide production and increased NOX2 subunit expression. In addition, endothelial function was significantly impaired, with both reduced ACh-mediated vasorelaxation and basal NO• bioactivity, as we and others have previously described.10,27,30
In addition, this study also showed that NaHS treatment could act to protect the activity of exogenously derived NO•. In diabetic aorta, the maximum relaxation response to the NO• donor SNP was significantly reduced, but treatment with NaHS restored the efficacy of exogenous NO• to control levels.
Previous work has shown that H2S is a KATP channel opener; 36 however, we saw no difference in the ex vivo aortic vasorelaxation response to the KATP channel opener LKM. These data show that the vascular smooth muscle cell function in diabetic mice was not compromised; thus, the decrease in vasorelaxation to ACh and SNP was due to a NO•-dependent deficit.
It is known that superoxide can readily react with and reduce the availability of NO•, 6 so it is possible that NaHS-induced reduction of NADPH oxidase activity and therefore superoxide production may be the mechanism behind this effect.
Recent studies have focused on the ability of H2S to elicit cytoprotective effects12,13 in a number of tissues by mechanisms that include potentially scavenging ROS,14,19 activating anti-oxidant gene expression (Nrf2 and nfKb) and enhancing expression of other anti-oxidant molecules.17,22 In addition, there is evidence that H2S may suppress ROS production by the NADPH oxidases. This study extends these findings of H2S-induced suppression of NADPH oxidase activity in the vasculature. There are seven isoforms of mammalian NADPH oxidase, which produce both superoxide and H2O2. These enzymes are essential for host defence and are important for cellular signalling; however, there is compelling evidence that overactivity of NADPH oxidase contributes to oxidative stress in cardiovascular diseases. 37
The effect of H2S on the various isoforms of NADPH oxidase has not been well studied. Previous studies in vascular smooth muscle cells showed that NaHS inhibited NADPH oxidase induced superoxide production, NOX1 subunit expression and Rac activity. 18 Another report showed NaHS could inhibit NADPH oxidase induced superoxide production and reduce NOX2 subunit expression in endothelial cells. 38 More recent in vivo studies have shown NaHS inhibits NOX4 expression in a rat model of myocardial ischaemia, resulting in reduced fibrosis 39 and reduced vascular NADPH oxidase activity has been shown in mouse models of angiotensin II–induced hypertension 23 and atherosclerosis. 24 This study has focused only on the NOX2 subunit of the enzyme as it has been previously shown to be pathologically up-regulated in cardiovascular diseases. 40 The data show that NaHS treatment could ameliorate the increase in NADPH-oxidase activity seen in diabetes, but there was no effect on NOX2 subunit expression, which is in contrast to the previous study, 38 the main difference being that this study was in aorta samples ex vivo rather than in cultured endothelial cells. It should be noted that in this study the superoxide signal was virtually abolished by DPI, a flavoprotein inhibitor, which is specific for NADPH oxidase at 5 µΜ, indicating that the superoxide detected in response to NADPH is derived from a NADPH oxidase. The data suggest that NaHS is reducing superoxide production by inhibiting NADPH oxidase activity, an effect that can occur in the absence of a decrease in NOX2 expression. It is possible that other subunits modulate the activity of NOX2-NADPH oxidase. Although unlikely it is not possible to exclude the possibility of NaHS inhibiting other ROS enzymatic sources in addition to NADPH oxidase. There is still a lack of information regarding H2S donors and their potential effects on NADPH oxidase subunits and more work in this area is warranted.
It is important to note that these results are obtained from a single daily ip injection of NaHS. NaHS is an inorganic salt which when dissolved releases H2S immediately; 41 thus, it is considered a ‘fast’ donor of H2S. The t1/2 of NaHS is estimated in the range of seconds to minutes, 42 then it is sequestered or metabolised to sulphate. Thus, the method of administration used in this study results in a transient high concentration of H2S which is quickly cleared as plasma sulphide levels are not different between the groups. This suggests the NaHS is having an immediate but long-lasting effect (e.g. via protein sulfhydration or altering gene expression), or that the H2S delivered is being sequestered or bound for later release. All these possibilities have been reported previously.17,22,43,44
In summary, chronic treatment with the fast H2S donor NaHS results in reduced oxidative stress in diabetes. This effect is possibly mediated via the inhibition of the NOX2-containing isoform of NADPH oxidase, which leads to a protection of the endogenous production and action of NO• in the vasculature. These data further confirm the anti-oxidant effect of H2S and H2S donors may prove to be a useful therapeutic in the treatment of diabetes-related vascular complications.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.