
Abstract
Objective:
Inhibitors of sodium-glucose cotransporter 2 ameliorate hyperglycaemia in diabetes by increasing urinary glucose excretion. However, the effects of sodium-glucose cotransporter 2 inhibitors on tubulointerstitial damage in diabetic nephropathy are not fully elucidated. We examined whether tofogliflozin, an inhibitor of sodium-glucose cotransporter 2, suppressed renal damage in KKAy/Ta mice, obese and type 2 diabetic animals.
Materials and methods:
Male 8-week-old KKAy/Ta mice or control C57BL/6J mice were kept on a standard diet with or without 0.015% tofogliflozin for 5 weeks. Blood glucose and blood pressure, body and kidney weight, urinary N-acetyl-β-
Results:
Although tofogliflozin treatment did not affect blood pressure, body weight or serum creatinine values, it improved hyperglycaemia and blocked the elevation of urinary N-acetyl-β-
Conclusion:
Our present results demonstrated that tofogliflozin could suppress albuminuria and tubulointerstitial injury in obese and type 2 diabetic mice. Inhibition of glucose entry into tubular cells by tofogliflozin may exert renoprotective properties in diabetes.
Introduction
According to the Diabetes Atlas, 7th edition, 2015, an estimated 415 million people worldwide had diabetes. 1 Among various vascular complications of diabetes, diabetic nephropathy is a global health challenge because it is a leading cause of end-stage renal disease, which could contribute to the increased risk of cardiovascular disease and death in diabetic patients. 2 Recently, inhibitors of sodium-glucose cotransporter 2 (SGLT2) have been developed as a novel oral hypoglycaemic agent. 2 Since 90% of glucose filtered by the glomerulus is reabsorbed by a low-affinity or high-capacity SGLT2 expressed on the apical membrane of proximal tubules at S1 and S2 segments, inhibitors of SGLT2 have a potential to improve hyperglycaemia in diabetic patients by promoting urinary glucose excretion. 2 Moreover, SGLT2 inhibitors have also been shown to ameliorate hypertension, hyperuricaemia and central obesity in patients with diabetes. 2 Since cardiometabolic derangements, including chronic hyperglycaemia and hypertension, are major risk factors for vascular complications in diabetes, treatment with SGLT2 inhibitors may be a promising strategy for preventing diabetic nephropathy. 2
Injury and apoptosis of proximal tubular cells have played a central role in the pathogenesis of tubular atrophy, tubulointerstitial fibrosis and atubular glomeruli, and these histological changes are most closely correlated with progression of renal dysfunction in diabetic patients.2–4 Furthermore, we have found that increased glucose entry into cultured renal proximal tubular cells via SGLT2 stimulates oxidative stress generation and subsequently evokes pro-apoptotic and pro-inflammatory reactions in this cell type. 5 These findings suggest that SGLT2 inhibitors may protect against tubulointerstitial injury in diabetes. However, the effects of SGLT2 inhibitors on tubulointerstitial damage in diabetic nephropathy are not fully elucidated. Therefore, we examined here whether tofogliflozin, a highly selective inhibitor of SGLT2, suppressed renal damage in KKAy/Ta mice, obese and type 2 diabetic animals.
Materials and methods
An oral selective inhibitor of SGLT2, tofogliflozin, was generously gifted from Sanofi K.K., Tokyo, Japan. Male 8-week-old KKAy/Ta mice or control C57BL/6J mice were purchased from CLEA Japan, Inc., Tokyo, Japan. Animals were kept on a standard MF-diet (Oriental Yeast Co., Ltd, Tokyo, Japan) with or without 0.015% tofogliflozin for 5 weeks. Animals at 9, 11 and 13 weeks old were housed in metabolic cages to collect urine for measurement of N-acetyl-β-
Results
As shown in Table 1, compared with control mice at 9, 11 and 13 weeks, body weight and blood glucose levels were significantly higher in KKAy/Ta diabetic mice at the same ages, respectively. Although tofogliflozin treatment for 5 weeks did not affect the increase in body weight of KKAy/Ta mice during the study periods, it improved hyperglycaemia in diabetic mice; tofogliflozin treatment significantly decreased blood glucose levels of KKAy/Ta mice at 9 and 13 weeks old, whereas blood glucose values of 11-week-old KKAy/Ta mice had a tendency to be decreased by tofogliflozin treatment.
Characteristics of animals.
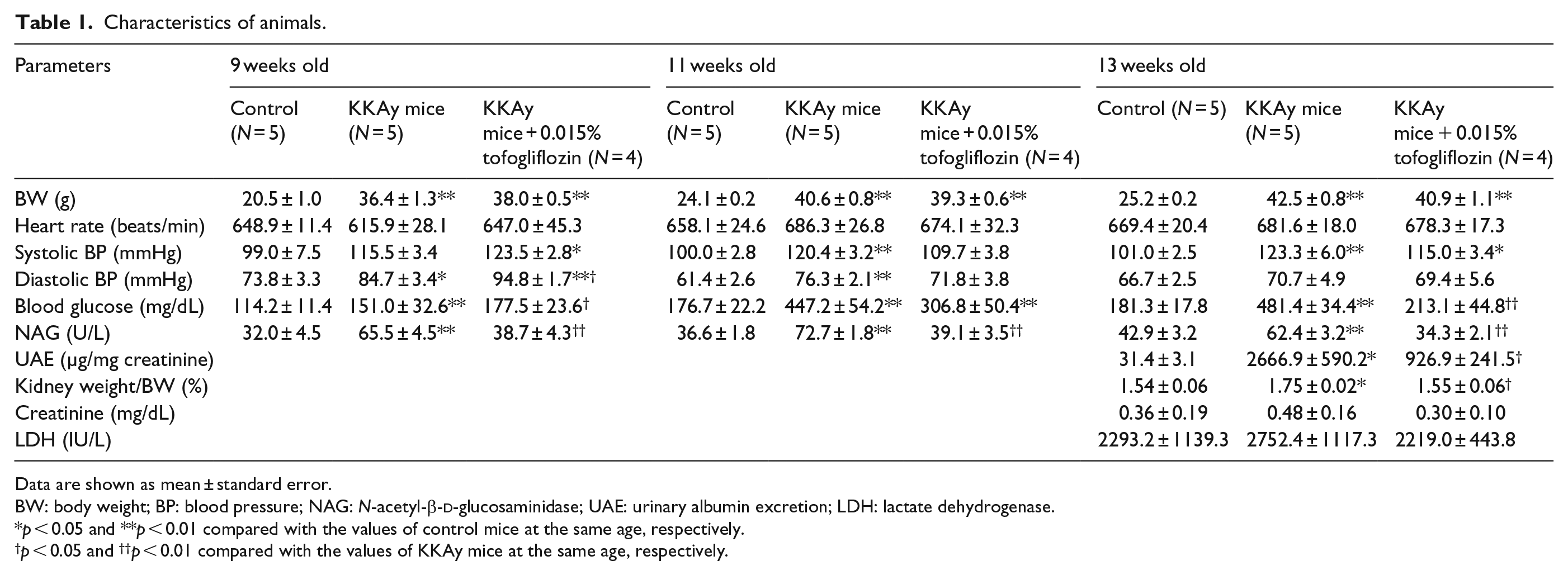
Data are shown as mean ± standard error.
BW: body weight; BP: blood pressure; NAG: N-acetyl-β-
p < 0.05 and **p < 0.01 compared with the values of control mice at the same age, respectively.
p < 0.05 and ††p < 0.01 compared with the values of KKAy mice at the same age, respectively.
Although systolic or diastolic BP of KKAy/Ta mice at 9, 11 or 13 weeks were significantly elevated compared with control mice at the same ages, tofogliflozin treatment did not affect BP of diabetic mice except for diastolic BP of 9-week-old KKAy/Ta mice. During the study periods, there was no significant difference of heart rate among the three groups. Serum creatinine or lactate dehydrogenase (LDH) levels at 13-week-old control mice, KKAy/Ta mice and tofogliflozin-treated KKAy/Ta mice did not differ.
NAG activities in the urine were significantly increased in KKAy/Ta diabetic mice at 9, 11 and 13 weeks compared with control mice at the same ages, respectively, all of which were reduced by the treatment with tofogliflozin (Table 1). Furthermore, tofogliflozin treatment for 5 weeks significantly inhibited the increases in UAE levels and kidney weight of KKAy/Ta diabetic mice at 13 weeks (Table 1).
Conclusion
In this study, we found for the first time that treatment with a standard diet containing 0.015% tofogliflozin significantly prevented the elevation of NAG activities in the urine of KKAy/Ta mice at 9, 11 and 13 weeks, an animal model of type 2 diabetes with obesity. We also found here that increases in UAE levels and kidney weight of KKAy/Ta diabetic mice at 13 weeks were significantly suppressed by the treatment with tofogliflozin. Urinary activity of NAG is one of the sensitive biomarkers for tubulointerstitial injury and dysfunction, 7 and its levels were reported to increase in type 2 diabetic patients with microalbuminuria compared to those with normoalbuminuria. 8 These observations indicate that tubulointerstitial damage is already present at early phase of diabetic nephropathy, thus suggesting the clinical utility of measurement of urinary NAG activity for early detection of diabetic nephropathy. Furthermore, urinary NAG activity has also been shown to be a useful marker for predicting renal prognosis in patients at early stage of diabetic nephropathy, whose prognostic value was lost after adjustment for interstitial fibrosis and tubular atrophy score. 8 These findings further support the concept that urinary NAG activity may be associated with histological damage within the tubulointerstitial areas, thereby being a therapeutic target for diabetic nephropathy. Therefore, this study suggests that tofogliflozin treatment may prevent the progression of diabetic nephropathy in obese type 2 diabetes by suppressing tubulointerstitial injury and reducing albuminuria. However, in this study, histological examination revealed no morphological alterations, such as tubular dilation, atrophy, glomerular sclerosis and tubulointerstitial fibrosis in KKAy/Ta mice at 13 weeks with a short duration of diabetes (data not shown). So, it would be interesting to examine whether tofogliflozin could prevent histological alterations within the glomerular and tubulointerstitial areas of KKAy/Ta mice with a longer history of diabetes.
Plasma-free tofogliflozin level is shown to be between 120 and 350 nM in mice after a consumption of diet containing 0.015% tofogliflozin. 9 In addition, the peak plasma concentration of tofogliflozin in humans after single oral dose of 20 mg is reported to be about 1 µM. 10 Since ca. 80% of tofogliflozin in the blood is a protein-bound form, its free level is estimated to be ~200 nM in humans. 10 Therefore, the applied dose of tofogliflozin used here is comparable to the therapeutic dose for diabetic patients.
In this study, we could not clarify the molecular mechanism how tofogliflozin treatment prevented tubulointerstitial damage in obese diabetic animals. However, we have previously found that inhibition or knockdown of SGLT2 not only inhibits glucose overload-induced oxidative stress generation, apoptotic cell death and monocyte chemoattractant protein-1 expression in proximal tubular cells partly by reducing expression of receptor for advanced glycation end products (RAGE), but also suppresses oxidative stress, tubulointerstitial damage and inflammatory and fibrotic reactions in the kidneys of type 1 diabetic rats by blocking the advanced glycation end products (AGEs)-RAGE axis.5,6,11 Therefore, tofogliflozin treatment may protect against tubulointerstitial injury of obese type 2 diabetic mice partly via suppression of glucose toxicity in tubular cells. In nested case–control of participants of the Diabetes Control and Complications Trial, although urinary excretion levels of AGEs predicted the progression of diabetic nephropathy, its predictive value was lost after adjustment for baseline urinary activity of NAG. 12 These observations suggest that increased urinary NAG activity is a marker that may reflect the abnormal tubular processing of AGEs or the AGEs-RAGE-induced tubulointerstitial damage in diabetes. Blockade of the SGLT2-mediated glucose uptake into tubular cells by tofogliflozin may inhibit the AGEs-RAGE axis in the diabetic kidney.
Footnotes
Acknowledgements
S. Yamagishi conceptualized and designed the study; acquired, analysed and interpreted data; and drafted this article, and he takes responsibility for the integrity of the data and the accuracy of the data analysis. Y. Ishibashi and T. Matsui acquired, analysed and interpreted data.
Declaration of conflicting interests
Dr Yamagishi has received honoraria such as lecture fees from Sanofi K.K., Tokyo, Japan.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research B (grant number 22390111; S.Y.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan and was financially supported in part by Sanofi K.K., Tokyo, Japan.