
Abstract
We have previously reported that advanced glycation end products activated Rho-associated protein kinase and p38 mitogen–activated protein kinase, causing endothelial hyperpermeability. However, the mechanisms involved were not fully clarified. Here, we explored the role of myosin light chain kinase in advanced glycation end product–induced endothelial hyperpermeability. Myosin light chain phosphorylation significantly increased by advanced glycation end products in endothelial cells in a time- and dose-dependent manner, indicating that myosin light chain phosphorylation is involved in the advanced glycation end product pathway. Advanced glycation end products also induced myosin phosphatase–targeting subunit 1 phosphorylation, and small interfering RNA knockdown of the receptor for advanced glycation end products, or blocking myosin light chain kinase with its inhibitor, ML-7, or small interfering RNA abated advanced glycation end product–induced myosin light chain phosphorylation. Advanced glycation end product–induced F-actin rearrangement and endothelial hyperpermeability were also diminished by inhibition of receptor for advanced glycation end product or myosin light chain kinase signalling. Moreover, inhibiting myosin light chain kinase with ML-7 or blocking receptor for advanced glycation end product with its neutralizing antibody attenuated advanced glycation end product–induced microvascular hyperpermeability. Our findings suggest a novel role for myosin light chain and myosin light chain kinase in advanced glycation end product–induced endothelial hyperpermeability.
Keywords
Introduction
Vascular and microvascular complications affect many organ systems and are the major reason of mortality and morbidity in patients with diabetes mellitus (DM). Increased vascular permeability due to impaired vascular endothelial cells structure and function is a major reason for vascular complications and a hallmark of DM.1,2 High intracellular glucose leads to the formation of advanced glycation end products (AGEs) via non-enzymatic glycosylation of proteins, which have been shown to cross-link proteins, accelerate atherosclerosis, reduce nitric oxide synthesis and induce endothelial dysfunction. 3 Receptor for advanced glycation end product (RAGE) is the major receptor for AGEs. Upon binding to RAGE, AGEs activate nuclear factor-κB (NF-κB), regulate the expression of proinflammatory cytokines and elicit inflammatory responses.4,5 We have previously demonstrated that AGEs caused the rearrangements of F-actin and endothelial hyperpermeability.6,7 However, the mechanisms by which AGEs increase endothelial permeability are not fully understood.
Myosin light chain kinases (MLCKs) belong to a family of soluble protein kinases that phosphorylate the regulatory myosin light chain (MLC)-2 to induce adenosine triphosphate (ATP)-dependent actomyosin contraction and thereby regulate arteriolar vasoconstriction and microvascular permeability. 8 Recent studies have reported that MLCK expression and MLC-2 phosphorylation were increased in DM, causing impaired systolic and diastolic function, and cytoskeletal rearrangements, leading to microvascular hyperpermeability.9,10 Therefore, we speculated that AGE-induced cytoskeletal rearrangements and endothelial hyperpermeability occurred via MLCK signalling. In this study, we investigated the role of MLCK and MLC signalling in AGE-induced endothelial hyperpermeability.
Methods
Materials and reagents
Human umbilical vein endothelial cells (HUVECs) were purchased from Sciencell (Carlsbad, CA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium was purchased from Hyclone (Logan, UT, USA). Foetal bovine serum and trypsin were purchased from Gibco BRL (Grand Island, NY, USA). Fluorescein isothiocyanate–labelled dextran (FITC-dextran), antibodies against MLC and phosphorylated MLC were purchased from Abcam (Cambridge, MA, USA). Small interfering RNA (siRNA) against RAGE and MLCK and a negative control siRNA (nonsense siRNA) were purchased from the Shanghai Gene Pharmar Corporation (Shanghai, China). Rhodamine-phalloidin was purchased from Molecular Probes (Eugene, OR, USA). The MLCK inhibitor, ML-7, was purchased from Santa Cruz (Santa Cruz, CA, USA). MLCK antibody was purchased from Sigma (St Louis, MO, USA). Myosin phosphatase–targeting subunit 1 (MYPT1) and phospho-specific MYPT1 (Thr853) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). RAGE neutralizing antibody (RAGE Ab) was prepared and characterized according to a previous study. 6
Preparation of AGE-modified bovine serum albumin
Advanced glycation end product–modified bovine serum albumin (AGE-BSA) was prepared as previously reported.
6
Briefly, BSA (150 mmol/L, pH 7.4) was incubated in phosphate-buffered saline (PBS) with
Cell culture and treatments
HUVECs were cultured in a humidified 5% CO2 incubator in DMEM/F12 containing 10% foetal calf serum (FCS), 100 U/mL penicillin and 100 µg/mL streptomycin for 2–3 days until they reached 90% confluence. The media were removed and cells were cultured in serum-free media for another 12 h before being used for the experiments. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) according to the manufacturer’s protocol for 48 h before being used in assays. In some experiments, cells were pretreated with 10 µmol/L ML-7 for 30 min, followed by 100 µg/mL AGE stimulation for the indicated times.
Western blotting
Western blotting was performed as previously described. 11 Total protein was extracted from cells and the concentration was determined with the Bradford assay. About 40 µg of samples was separated by 10% polyacrylamide gel electrophoresis and transferred to membranes. The membranes were blocked with Tris-buffered saline containing 0.1% Tween20 (TBST) and 5% non-fat milk at room temperature (RT) for 1 h and then probed with the appropriate primary antibody (1:1000) overnight at 4°C. After washing with TBST (10 min for four times), membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000) at RT for 1 h and washed in TBST (10 min for four times). Blots were exposed to ECL Plus (Amersham Biosciences, Pittsburgh, PA, USA) to visualize protein bands.
Monolayer endothelial cell permeability measurement
Monolayer endothelial cell permeability was measured as described previously. 12 HUVECs (105 cells/cm2) were seeded in a double-transparent dish (Corning Costar Corporation, Acton, MA, USA) covered with 1% gelatin for 2–3 days until they reached confluence, followed by incubation in serum-free media for another 12 h. Cells were then incubated in PBS with or without 100 µg/mL AGEs for 8 h. FITC-dextran (200 µL) at 1 mg/mL was added into the upper chamber for 45 min. One hundred microliters of sample was taken from both the upper and bottom chambers and assayed in a 96-well plate. Fluorescence intensity was read using a HTS 7000 microplate reader (Perkin-Elmer, Yokohama, Japan). The permeability coefficient for albumin (Pa) was calculated as follows: Pa = [A]/t × 1/A × V/[L], where [A] is the albumin concentration in bottom chamber, t is the time (s), A is the area of the membrane (cm2), V is the volume of the bottom chamber and [L] is the albumin concentration of upper chamber.
Immunofluorescent analysis
The media were removed and cells were rinsed with PBS, fixed in 4% paraformaldehyde at 4°C for l0 min, rinsed with PBS for 2 min for three times, incubated in 0.5% Triton X-100 at 4°C for 10 min and rinsed with PBS for 2 min for three times. Cells were then stained with 2 U/mL rhodamine-phalloidin in a dark box at RT for 1 h and rinsed with PBS for 2 min for three times. Fluorescent images were captured with a laser scanning confocal microscope (Leica TCS2SP2, Wetzlar, Germany).
Permeability detection in isolated venules
Sprague-Dawley rats, weighing160–200 g, were obtained from the Laboratory Animal Center of Southern China. All study protocols were approved by the Animal Care Committee of Southern Medical University of China, and the study was performed strictly according to the National Institute of Health guidelines. The permeability of isolated venules was determined according to a previous study. 13 Animals were injected intraperitoneally with AGEs (10 mg/kg) 14 and/or the inhibitor of MLCK, ML-7 (1 mg/kg), 15 and anaesthetized with a combination of ketamine (75 mg/kg) and midazolam (2.5 mg/kg) 6 h later. A suitable mesentery (diameter 60–0 µm, length 1.0–1.2 mm, no branch) was excised, immediately transferred into a cold albumin-physiological salt solution (APSS) separate pool and then mounted to a Zeiss microscope. The isolated venules were cannulated at each end with micropipettes and secured with an 11-0 suture, with the inflow pipette inserted with a smaller pipette. APSS or APSS containing FITC-labelled albumin was perfused from the inflow end of the venules. The permeability of the venules was quantified by measuring the ratio of the transvascular flux to the transmural concentration difference in albumin. Fluorescence video microscopy (Nikon ECLIPSE 80i, Tokyo, Japan) was used to monitor leaks and to perform experimental measurements within a selected field of view containing a venule of known size. The permeability coefficient (Pa) was calculated as follows: Pa = (1/ΔIf) × (dIf/dt)0 × (r/2),where ΔIf is the change in fluorescence intensity, (dIf/dt) represents the exudation velocity of fluorescent albumin and r is the radius of the venule. Data are expressed as the percentage of change relative to the control.
Statistical analysis
The data are presented as mean ± standard deviation (SD) of four to six independent experiments. Statistical analysis was performed using SPSS (13.0) software. p Values were calculated with one-way analysis of variance for multiple comparisons; p <0.05 was considered statistically significant.
Results
AGEs induce MLC phosphorylation in endothelial cells
To determine the effect of AGEs on MLC phosphorylation, HUVECs were incubated with AGE-BSA or BSA at 100 µg/mL for different durations or at different concentrations for 8 h, and the phosphorylation levels of MLC were determined by western blotting. Our data showed that the phosphorylation levels of MLC gradually increased by 100 µg/mL AGE-BSA, but not by BSA, and the increases were significant at 4 and 8 h (Figures 1(a) and Supplementary Figure 1A), indicating that AGEs induced MLC phosphorylation in a time-dependent manner. We also observed a concentration effect of AGE-BSA on MLC phosphorylation (Figures 1(b) and Supplementary Figure 1B). These data indicated that MLC phosphorylation may be involved in the AGE signalling pathway.
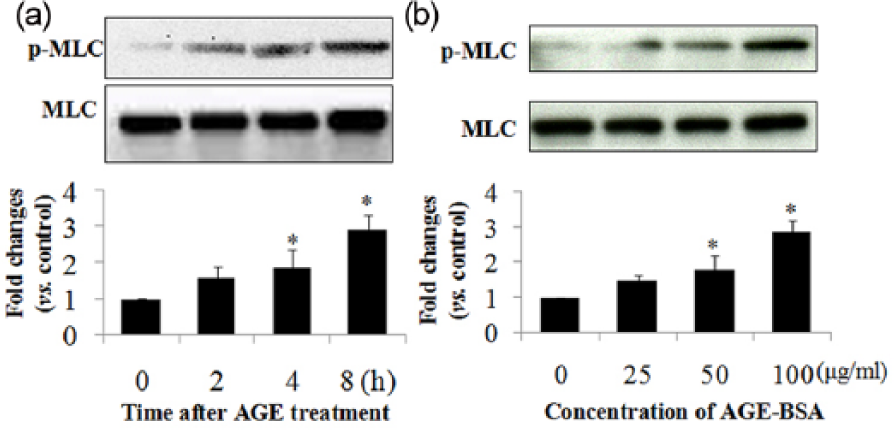
AGEs induce MLC phosphorylation in a time-dependent manner. Cells were incubated (a) with 100 µg/mL AGEs for 0, 2, 4 and 8 h or (b) with different concentrations of AGEs (0, 25, 50 and 100 µg/mL) for 8 h, and total protein was extracted. MLC and phosphorylated MLC were determined by western blotting. Data represent mean ± SD of four independent experiments. *p < 0.05, vs control.
Knockdown of RAGE downregulates MLC phosphorylation
As RAGE is the major receptor for AGEs and can be induced by AGE-BSA,16,17 we next examined whether knockdown of RAGE with siRNA influences AGE-induced MLC phosphorylation. HUVECs were transfected with RAGE siRNA (siRAGE) or the negative control (siCON) for 48 h, and then the effect of knockdown of RAGE protein on MLC phosphorylation was determined. We found that siRAGE decreased the protein expression of RAGE by about 70%. Moreover, the phosphorylation levels of MLC in HUVECs transfected with siCON did not change. AGEs significantly increased MLC phosphorylation in PBS or siCON-treated cells, and this increase was markedly suppressed by siRAGE (Figure 2(a) and (b)).These data suggest that MLC phosphorylation was induced mainly by AGEs binding to RAGE.
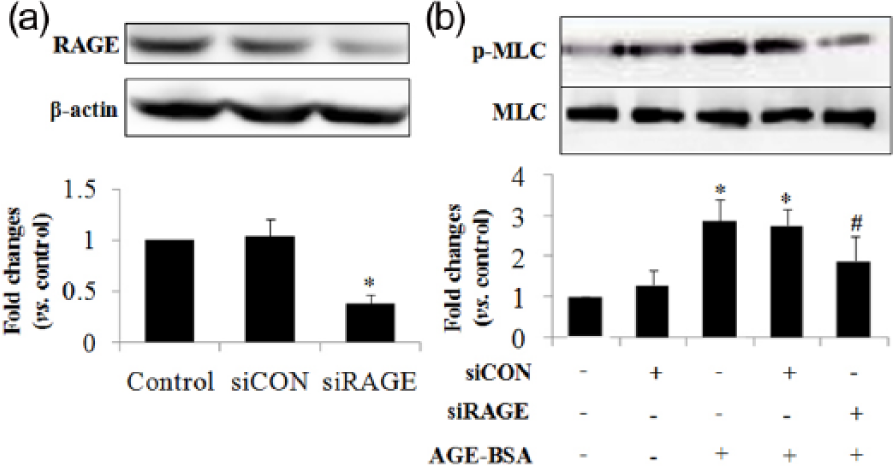
Knockdown of RAGE with siRNA decreased MLC phosphorylation levels in AGE-treated HUVECs. Cells were transfected with control siRNA or RAGE siRNA for 48 h. (a) RAGE protein expression was determined by western blotting. (b) After incubation with AGEs (100 µg/mL) or PBS for 8 h, MLC and phosphorylated MLC levels were determined by western blotting. Data represent the mean ± SD of four independent experiments. p < 0.05, *versus the corresponding control, #versus the AGEs group.
The role of MLCK and MLC phosphatase signalling in AGE-induced MLC phosphorylation
Given that MLC is mainly phosphorylated by MLCK and dephosphorylated by myosin light chain kinase phosphatase (MLCP), we investigated whether MLCK or MLCP is involved in the regulation of MLC phosphorylation. MYPT1 is the major subunit of MLCP and its phosphorylation leads to MLCP inhibition. We found that MYPT1 phosphorylation increased by AGE-BSA, indicating that MLCP inhibition is implicated in MLC phosphorylation (Figure 3(a)).
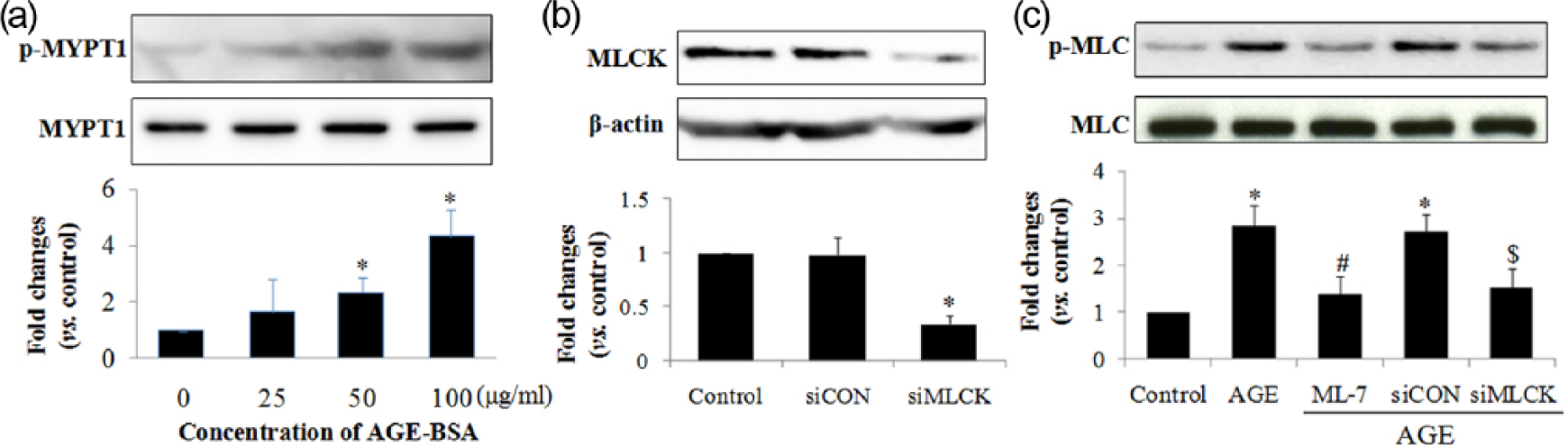
AGEs induce MYPT1 phosphorylation, and blocking MLCK with ML-7 or siRNA decreases MLC phosphorylation levels in AGE-treated HUVECs. (a) Cells were incubated with different concentrations of AGE-BSA (0, 25, 50 and 100 µg/mL) for 4 h, and MYPT1 and phosphorylated MYPT1 levels were evaluated by western blotting. (b) Cells were transfected with control siRNA or RAGE siRNA for 48 h, followed by incubation with AGEs (100 µg/mL), ML-7 (10 µmol/L) or PBS for 8 h. MLC and phosphorylated MLC levels were determined by western blotting. Data represent the mean ± SD of four independent experiments. p < 0.05, *versus the corresponding control, #versus the AGEs group, $versus the siCON group.
We next explored the role of MLCK in MLC phosphorylation. Cells were transfected with MLCK siRNA (siMLCK) for 48 h, or pretreated with 10 µmol/L ML-7 before being subjected to AGE stimulation for 8 h, and MLC phosphorylation was determined. We found that siMLCK decreased MLCK protein expression by nearly 75%, and that MLCK inhibition by siMLCK or ML-7 significantly inhibited AGE-induced MLC phosphorylation, suggesting that MLCK signalling contributes to MLC phosphorylation in AGE-treated endothelial cells (Figure 3(b) and (c)).
Blocking the RAGE-MLC pathway attenuates AGE-induced endothelial hyperpermeability
Because our findings suggest that AGEs induced MLC phosphorylation via RAGE and MLCK, we asked whether these proteins affect endothelial hyperpermeability induced by AGEs. Cells were transfected with RAGE or MLCK siRNA, or pretreated with ML-7, and monolayer endothelial cell permeability was determined using FITC-dextran. Our data showed that the endothelial permeability significantly increased in the AGE-treated control and the control siRNA-transfected groups. The increased endothelial permeability was markedly diminished by depletion of RAGE by siRNA, as well as by inhibition of MLCK by siRNA or its inhibitor, ML-7, suggesting that RAGE and MLCK mediated endothelial hyperpermeability in AGE-treated HUVECs (Figure 4).
Inhibition of RAGE or MLCK signalling decreases endothelial permeability in AGE-treated HUVECs. Cells were cultured in transwells and transfected with control siRNA or RAGE siRNA for 48 h, followed by incubation with AGEs (100 µg/mL), ML-7 (10 µmol/L) or PBS for 8 h. MLC and phosphorylated MLC levels were determined by western blotting. Data represent mean ± SD of four independent experiments. p < 0.05, *versus the corresponding control, #versus the AGEs group, $versus the siCON group.
Blocking RAGE and MLCK inhibits AGE-induced F-actin rearrangements
We have previously reported that the AGE-induced F-actin rearrangement was linked to increased endothelial permeability. Here, we asked whether RAGE and MLCK influenced the F-actin rearrangement. We observed that in the control group F-actin was mainly distributed in the periphery of the cells, with continuous intact lines and a typical outline of cobble stone, and the intercellular connections were tight without gap formation. However, in the AGE-treated group, F-actin at the outer peripheral edge of the cells was dense and rough, with irregular jagged-like fractures tending to dissipate and disintegrate and became dispersed with the formation of stress fibres in the cytoplasm. The F-actin rearrangements were inhibited by RAGE knockdown, or by MLCK inhibition by its inhibitor, ML-7, or by siRNA (Figure 5). These data suggest that RAGE and MLCK are important for AGE-induced F-actin rearrangements.
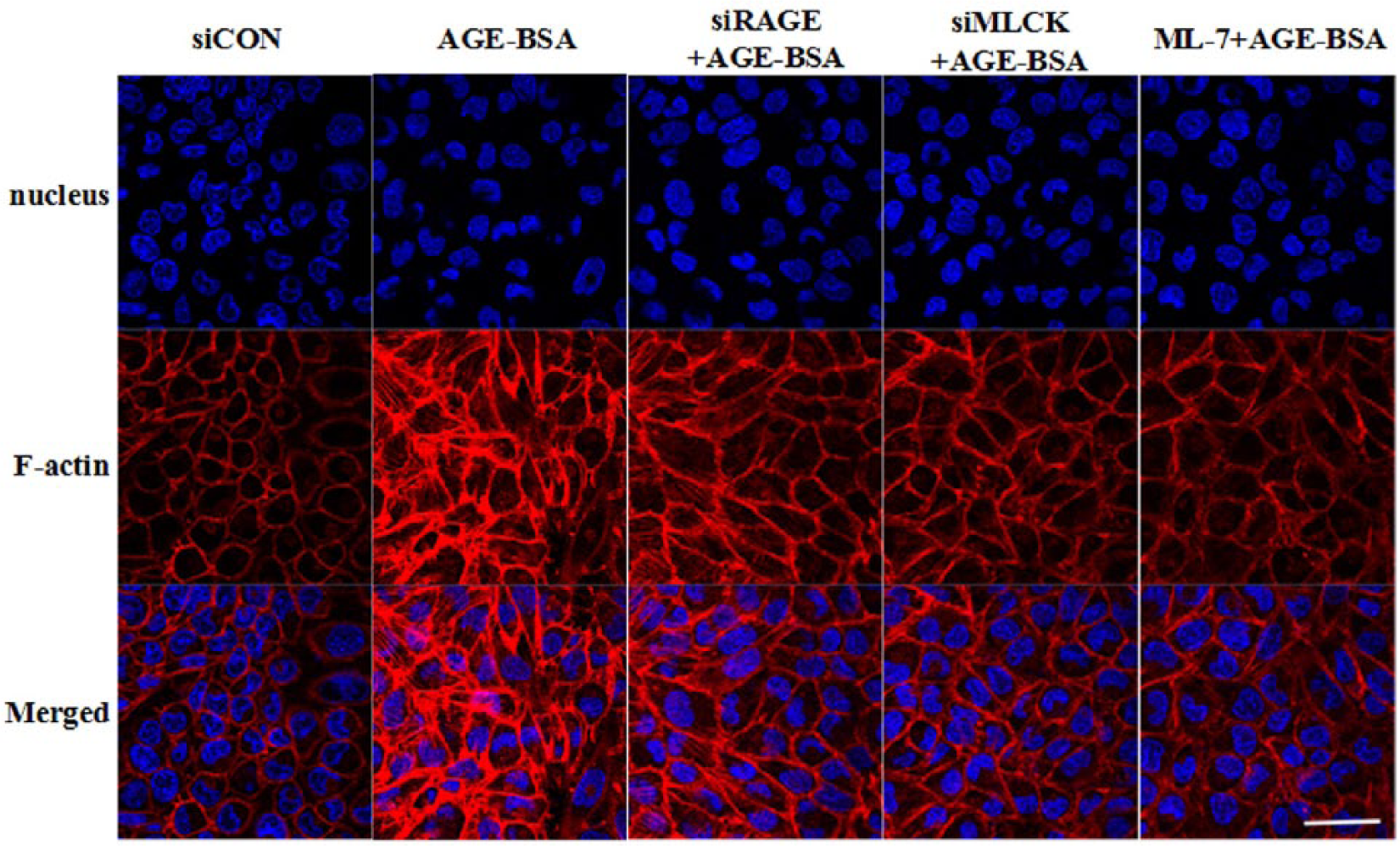
Inhibition of RAGE or MLCK signalling diminishes F-actin rearrangements in AGE-treated HUVECs. Cells were cultured in Petri dish and transfected with control siRNA or RAGE siRNA for 48 h, followed by incubation with AGEs (100 µg/mL), ML-7 (10 µmol/L) or PBS for 8 h. MLC and phosphorylated MLC levels were determined by western blotting. Representative images are shown. Scale bar: 50 µm.
Inhibition of MLCK or RAGE diminishes AGE-induced hyperpermeability in isolated venules
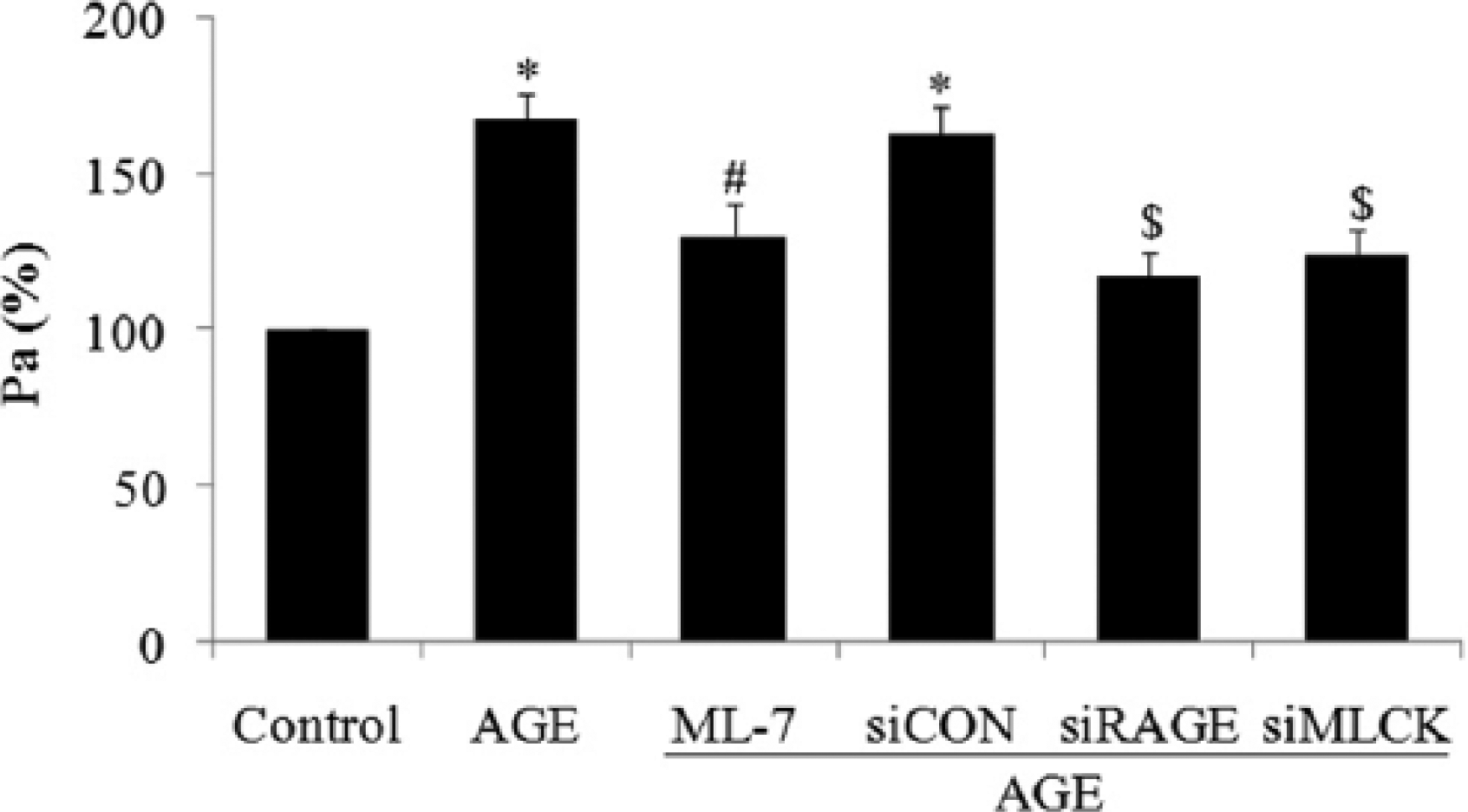
We next explored the effects of MLCK or RAGE on microvascular permeability in AGE-treated rats. Isolated venules were perfused with FITC-labelled albumin. Pa values were calculated to evaluate venule permeability. Our data showed that AGEs significantly increased the exudation of FITC-labelled albumin from venules, which was remarkably attenuated by ML-7 or RAGE antibody. These data further proved the role of MLCK in AGE-induced endothelial hyperpermeability (Figure 6).
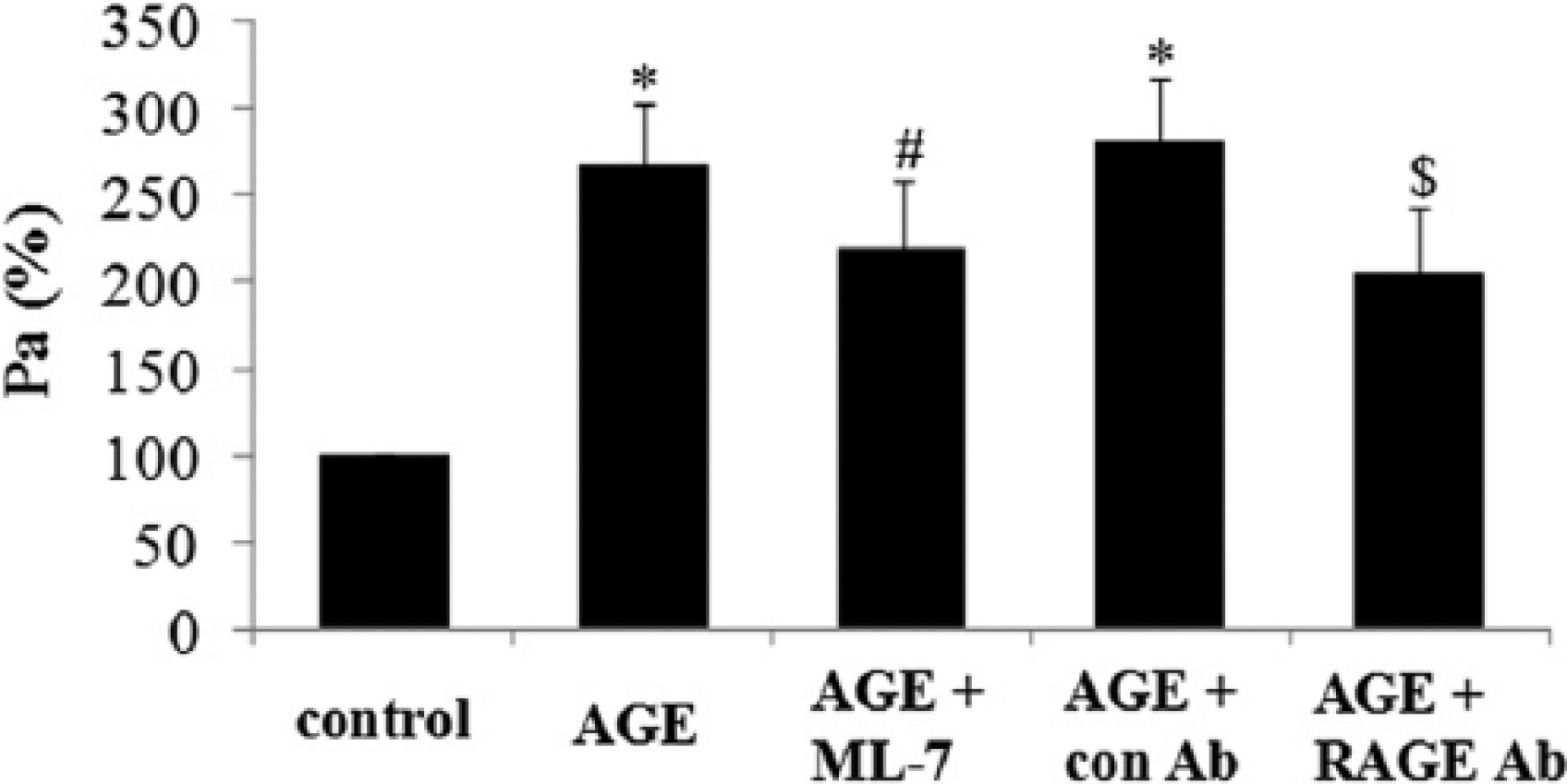
Inhibition of MLCK with ML-7 decreases microvascular permeability in AGE-treated rats. Rats were injected intraperitoneally with BSA (10 mg/kg; control), AGEs (10 mg/kg), RAGE neutralizing antibody (RAGE Ab) or ML-7 (1 mg/kg) for 8 h, followed by anaesthesia. Mesentery venules were isolated and perfused with FITC-albumin. Fluorescent images were captured and the permeability coefficient (Pa) was calculated. p < 0.05, *versus the corresponding control, #versus the AGEs group, $versus the AGE + con Ab group; n = 5.
Discussion
We have previously shown that AGEs activated Rho-associated protein kinase (ROCK) and the p38 mitogen–activated protein kinase pathway, and then phosphorylated moesin, resulting in F-actin rearrangements and endothelial hyperpermeability. 6 However, the mechanisms are not fully understood. Studies have shown that MLCK activation and MLC phosphorylation mediate disruption of the barrier function in endothelial cells subjected to various extracellular stimuli, such as cytokines and reactive oxygen species;10,18 however, their role in AGE-induced endothelial hyperpermeability remains unclear. Moreover, it has been reported that MLCK is inhibited by adenosine monophosphate (AMP)-activated protein kinase (AMPK) and the inhibition of RAGE attenuates AMPK signalling in smooth muscle cells (SMCs),19–21 which indicates that AGEs inhibit rather than activate MLCK via the RAGE-AMPK signalling pathway. Thus, it is necessary to investigate the role of MLCK signalling in AGE-treated endothelial cells. This study demonstrated that MLC has a role in AGE-induced endothelial hyperpermeability. AGEs have been reported to induce F-actin rearrangements and increase endothelial and microvascular permeability. Here, we found that AGEs increased MLC phosphorylation in a time-dependent manner and the inhibition of AGEs signalling by siRAGE diminished this phosphorylation, suggesting that MLC phosphorylation is involved in the AGE signalling pathway. Because MLC is mainly phosphorylated by MLCK and in this study MLC phosphorylation was suppressed by MLCK inhibition, we inhibited MLCK with its inhibitor or with siRNA and found that AGE-induced endothelial hyperpermeability and F-actin rearrangements were also attenuated by MLCK inhibition. Similar results were obtained when inhibiting RAGE with its siRNA. These data suggest a novel mechanism by which upon binding to RAGE, AGEs induce endothelial hyperpermeability via MLC phosphorylation. Importantly, we confirmed the role of MLCK in vivo, suggesting that MLC and MLCK are potential molecular targets of vascular complications in DM.
Previous studies have shown that MLCK activation and MLC phosphorylation were involved in endothelial hyperpermeability. The basis of the endothelial barrier consists of intercellular junctions, mainly adherens junctions (AJs), which are regulated by cytoskeletal contraction. Enhanced actomyosin contractility or stress fibres formed by F-actin rearrangements in response to various extracellular stimuli or intracellular signalling increase the inward tension, initiate endothelial cell contraction and disrupt the endothelial barrier. It has also been reported that MLCK activation by Ca2+/calmodulin or tyrosine kinase phosphorylated MLC at Ser19 and subsequently at Thr18, 22 increasing actomyosin contractility and resulting in endothelial hyperpermeability. On the contrary, MLCP dephosphorylates MLC, decreasing actomyosin contractility, relaxing the cytoskeleton and reducing paracellular permeability. 23 Furthermore, MLCP is phosphorylated and inactivated by ROCK, causing increased MLC phosphorylation and endothelial hyperpermeability. 24 This process is MLCK dependent, but does not require elevated MLCK activity. Given that AGEs induce ROCK activation, 6 we speculated that the increased MLC phosphorylation by AGEs resulted from ROCK-mediated inhibition of MLCP in an MLCK-dependent manner, which is also supported by our findings that AGEs inhibited MLCP by phosphorylating MYPT1. Other mechanisms may also be implicated in AGE-induced MLC phosphorylation. For example, AGEs can induce MLC phosphorylation via Ca2+ because the intracellular Ca2+ levels increase in response to AGE stimulation, forming a Ca2+–calmodulin complex that activates MLCK, causing MLC phosphorylation.18,25,26 AGE-induced MLC phosphorylation may also be mediated by Src signalling given that Src activation is involved in MLCK phosphorylation, and the Src kinase can be activated by RAGE and inhibited by the specific RAGE antibody in vascular smooth muscle cells (VSMCs).27,28
Importantly, we tested the role of MLCK in vivo with its inhibitor, ML-7, and found that AGE-induced microvascular hyperpermeability was alleviated by ML-7, which is consistent with our observation in HUVECs. Moreover, as SMCs are also involved in the regulation of microvascular permeability and the MLCK isoform in SMCs phosphorylates MLC, causing SMC contraction and increased microvascular permeability, the protective effects against AGE-induced microvascular hyperpermeability exerted by ML-7 may be linked to the inhibition of SMC contraction. 18 Given that as the major cause of microvascular complications, AGEs induce microvascular hyperpermeability and cause tissue oedema and organ dysfunction, 29 our data suggest that MLC and MLCK are potential targets for the treatment of vascular complications associated with DM.
In summary, our findings suggest that AGEs induce endothelial hyperpermeability via the MLC pathway, providing a novel insight into the treatment of microvascular complications in DM. However, there are limitations to our study. We blocked MLCK with its inhibitor or siRNA, which may have off-target effects. Thus, it is necessary to confirm the in vivo role of MLCK by gene-specific knockout methods.
Footnotes
Author contribution
F.W. and X.G. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
This work was supported by grants from the PhD Start-up Fund of Natural Science Foundation of Guangdong Province, China (S2013040015661); China Postdoctoral Science Foundation (2014 M552180); National Natural Science Foundation of China (31300950); Natural Science Foundation of Guangdong Province (2014A030313301); Project of Medical Research of PLA (BWS12J108) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.