
Abstract
Pro-inflammatory conditions induced by products of protein glycation in diabetes substantially enhance the risk of endothelial dysfunction and related vascular complications. Endothelial cell specific molecule-1 (ESM-1) or endocan has been demonstrated as a potential biomarker in cancer and sepsis. Its role in diabetes-induced pathologies remains unknown. The expression of ESM-1 gene is under cytokine regulation, indicating its role in endothelium-dependent pathological disorders. In this study, we investigated the effect of advanced glycated human serum albumin (AGE-HSA) on the production of ESM-1. We show that AGE-HSA exerts a modulating role on the expression of ESM-1 in human umbilical vein endothelial cells. It up-regulates expression of ESM-1 protein in a dose-dependent manner which correlates with its messenger RNA (mRNA) transcription. RAGE and galectin-3, both AGE receptors, show antagonistic action on its expression. While gene silencing of RAGE has down-regulatory effect, that of galectin-3 has up-regulatory effect on AGE-induced expression of ESM-1. Inhibition of MAPKKK and JNK pathways did not alter the expression. In contrast, phosphatidylinositol 3 kinase (PI3K) inhibition significantly up-regulated ESM-1 expression. In conclusion, these results suggest that AGE-induced activation of human umbilical vein endothelial cells promotes formation of endocan which is an endothelial dysfunction marker and may be related to vascular disease in diabetes.
Keywords
Introduction
Advanced glycated end products (AGEs) are reactive derivatives of non-enzymatic glycation reactions between glucose and proteins and mediate pro-inflammatory conditions in multiple ways. The direct effect of AGEs on vascular functions is partly unknown even though it is recognized that development of AGE-induced diabetic vascular injury is manifested through their engagement with corresponding receptors. Several AGE-binding receptors have been identified and characterized like RAGE (35-kDa), a member of the immunoglobulin superfamily, 1 the macrophage scavenger receptor classes A 2 and B, 3 the 60- and 90-kDa proteins AGE receptor 1 and 2 (AGE-R1 and AGE-R2), respectively, and galectin-3 or AGE-R3 (32-kDa),4,5 and almost all are expressed in endothelial cells. While AGE–RAGE interactions are widely reported, the other receptors have received less attention. Studies have demonstrated the presence and location of galectin-3 in different cell types, such as macrophages, eosinophils, neutrophils, mast cells, epithelia of respiratory and digestive tracts, some sensory neurons, and fibroblasts.6,7 It is mainly present in the cytosol but may be secreted extracellularly, where it binds to cell surface glycoconjugates exhibiting beta-galactosides. 8 Galectin-3 and its binding with AGE ligands have been detailed in macrophage cell line. 5
Endothelial cell specific molecule-1 (ESM-1) also known as endocan is constitutively expressed in the human lung and kidney endothelial cells 9 and is under vascular endothelial growth factor (VEGF) regulation. It has been established as a biomarker in cancer and sepsis,10,11 and inflammatory cytokines and pro-angiogenic growth factors increase its expression. Although it has not been found to be specific for any systemic inflammatory diseases, it is known to mediate leukocyte adhesion and activation. 12 Its role in patho-physiology of diabetes is unknown, except in proliferative diabetic retinopathy (PDR) where levels of endocan were found to be significantly high in vitreous fluid in PDR patients as compared to normo-glycaemic individuals. 13 Its role in AGE-mediated pro-inflammatory conditions is yet to be demonstrated. Since the expression of endocan gene is under the regulation of cytokines, it is likely to participate in endothelium-dependent pathological disorders.
In an effort to gain an understanding of how AGEs may modulate the expression of the endothelial biomarker endocan in the context of diabetic vascular injury and the effect of RAGE and galectin-3 AGE receptors, on its expression, we have exposed human umbilical vein endothelial cells (HUVECs) to 12- and 20-mM glucose-derived glycated human serum albumin (AGE-HSA). This concentration of glucose corresponds to blood glucose levels of 216 and 360 mg/dL (moderate and poorly controlled hyperglycaemia). Expression of endocan was correlated with ESM-1 messenger RNA (mRNA), and mechanistic pathways involved were also examined.
Experimental methods
Isolation of endothelial cells in vitro
Human umbilical cords were collected from Central Government Health Scheme (CGHS) maternity and gynaecology hospital (New Delhi, India) after obtaining informed consent from the patients. Investigation conforms to the principles outlined in the Declaration of Helsinki. As per Indian Council of Medical Research guidelines, studies involving unidentifiable specimens obtained from the provider, that is prohibited from releasing identifiers by established regulations, qualify for exempt status. HUVECs were isolated as per the previously described method. 14 Cells were grown in endothelial cell growth media-2 (EGM-2; Promocell, Germany) containing growth supplements. Cells were cultured in T25 cell culture flasks coated with gelatin and fibronectin (0.2% gelatin in phosphate buffered saline (PBS) pH 7.4 and human fibronectin 2 µg/cm2) and maintained at 37°C in a humidified CO2 incubator (MMM Medcenter Einrichtungen, Germany). HUVECs were characterized by their typical cobblestone appearance (phase contrast) and by uptake of DIL-Ac-LDL 15 visualized under confocal microscope (Olympus FLOW VEIW FV 1000 with camera DP-71; Japan; 40× objectives).
Exposure of cultured cells to hyperglycaemic conditions
Third passage HUVECs cultured to confluence in 60-mm culture plates and pre-coated with gelatin and fibronectin were used for the experiment. Following removal of media and washing with Hank’s Balanced Salt Solution (HBSS), cells were serum-starved overnight in preparation for AGE-HSA treatment. AGE-HSA was prepared by incubating a physiological concentration of HSA (40 g/L) with two different concentrations of glucose solution 12 mM (216 mg/dL; moderate hyperglycaemia) and 20 mM (360 mg/dL; poorly controlled hyperglycaemia). 16 Cells were incubated with AGE-HSA (2 mg/mL) derived from 12-mM or 20-mM glucose for 4 h at 37°C. A control plate without any treatment was also incubated under similar condition. Protein expression of RAGE, ESM-1 and galectin-3 was quantified immediately after exposure by standard enzyme-linked immunosorbent assay (ELISA) method as per the protocol mentioned in the kit (Raybiotech Inc., USA; Cusabio Biotech, China). It was ensured through the cell viability (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (MTT) assay that the metabolic activity of exposed HUVECs did not alter significantly under given experimental conditions.
RNA isolation and quantitative real-time polymerase chain reaction
Total messenger RNA (mRNA) was isolated from control and treated cells using RNeasy mini kit (Qiagen, USA). RNA concentration and purity were determined photometrically (Perkin Elmer, USA). Complementary DNA (cDNA) was synthesized from the template mRNA by using QuantiTect Reverse Transcription Kit (Qiagen). Quantitative real-time polymerase chain reaction (PCR) was performed using commercially available real time PCR kit of Qiagen (QuantiTect® Reverse Transcription; Qiagen) in Bio-Rad LightCycler (USA). Reactions were carried out in 25 µL reaction mixture containing 1 µL cDNA, 1.25 µL probes, 12.5 µL of 2× master mix and volume made up with RNA-grade double distilled water. Sequence-specific probes for the assay were designed and synthesized (Assays on Demand, ESM-1 QuantiFast duplex probes (QF00439817); Qiagen). The relative expression levels of gene of interest were calculated using the 2-(Δct) method with β-actin as control (house keeping gene).
RAGE and galectin-3 signalling in regulation of ESM-1 in HUVEC
The potential effect of AGE/RAGE binding and AGE sequestration by galectin-3 binding on regulation of ESM-1 was determined with RAGE and galectin-3 gene silencing. HUVECs grown in six-well plates to ~80% confluence were transfected with the siRNA duplex (final concentration 75 µg/mL) using transfection medium. The 6-h post-transfected cells were then grown for further 24 h in growth medium supplemented with 2× serum and antibiotics. Following this, they were treated with 2 mg/mL AGE-HSA for 4 h. The target siRNA for RAGE and galectin-3 and a negative-control siRNA with an irrelevant sequence were purchased from Santa Cruz Biotechnology, USA. Expression of ESM-1 was assessed by ELISA as mentioned before.
Regulation of ESM-1 transcription by intracellular signalling pathways
In another series of experiments, effects of mitogen-activated protein kinase (MAPK) pathway inhibition on regulation of ESM-1 production in HUVECs were examined by culturing cells with inhibitors MAPKKK Activation Inhibitor (10 µM; 570100-5MGCN) and JNK inhibitor II [10 µM; 420119-5MGCN (Calbiochem; Merck, Germany)]. The intracellular signalling pathway mediated by phosphatidylinositol 3 kinase (PI3K) was also checked with PI3Kd inhibitor (10 µM; 526559-5MGCN). After treatment, expression of ESM-1 protein was determined by ELISA and compared with control.
Data analysis
Data were expressed as the mean ± standard deviation (SD). Statistical two-group comparisons were performed by paired Student’s t-test.
Results
Metabolic stress has been evaluated in terms of glycated serum albumin. The process of glycation follows the time course of early, intermediate and late stages. 17 The terminal products are yellow-brown and fluorescent irreversible AGEs. Monitoring of the fluorescence gives an idea about the progress of the non-enzymatic glycation leading to the final product. Glycation of HSA with 12- and 20-mM glucose for a period of 14 weeks produced a heterogeneous group of glycated species which was verified by increase in molecular weight through matrix-assisted laser desorption/ionization (MALDI) analysis. The higher molecular weight of the protein and enhanced fluorescence intensity achieved with 20-mM glucose indicates more glycation compared to 12-mM glucose (data not shown). Cell viability under different experimental treatments was found to be around 95% as checked with MTT assay.
AGE-HSA promotes expression of ESM-1 protein in a dose-dependent manner in HUVECs
We first investigated the effect of AGE-HSA derived from 12- and 20-mM glucose (moderate and poorly controlled hyperglycaemia, respectively) on ESM-1 expression. After reaching confluence, cells were treated with EGM-2 growth media containing 2 mg/mL of AGE-HSA for 4 h. Whole cell lysates were analysed by ELISA. In control cells, expression of ESM-1 was detectable which was up-regulated at both levels of AGE-HSA. A significant increase by 35% was observed only in the presence of 20-mM glucose-derived AGE-HSA (Figure 1, p < 0.0001). Next, we examined whether the increase in ESM-1 protein levels induced by AGE-HSA is concomitant to increase at the transcriptional level. Total RNA was analysed by real time polymerase chain reaction using β-actin as internal control. AGE-HSA was found to modify ESM-1 RNA levels in the presence of both 12- and 20-mM glucose-derived AGE-HSA. A significant increase by 47% was observed only in the presence of 20-mM glucose-derived AGE-HSA (Figure 1, p < 0.01).
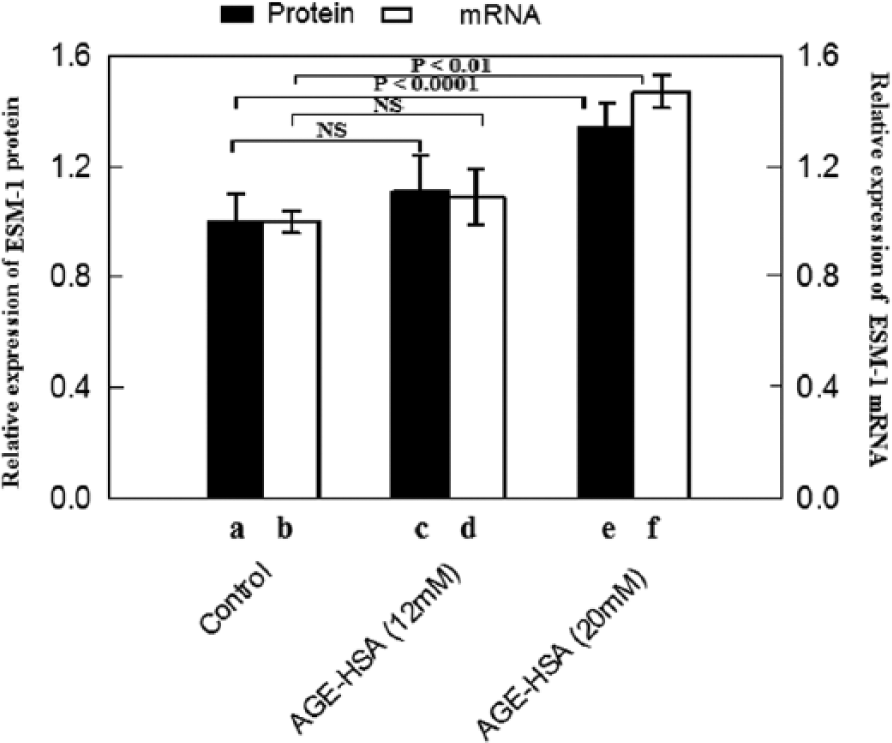
Effect of 12- and 20-mM glucose-derived AGE-HSA (2 mg/mL) on the relative (to control) expression of ESM-1 protein (n = 6) and mRNA (n = 3) in HUVECs. (a), (b) Control; (c), (d) 12-mM glucose-derived AGE-HSA; and (e), (f) 20-mM glucose-derived AGE-HSA. Data are expressed as mean ± standard deviation (paired t-test; p < 0.05 was considered significant).
Furthermore, to understand the role of receptors for AGEs in AGE-HSA-induced up-regulation of ESM-1, we first evaluated the RAGE and galectin-3 response of endothelial cells to AGE-HSA. Expression of RAGE and galectin-3 was measured in the cells in the presence of 12- and 20-mM glucose-derived AGE-HSA at 2 mg/mL concentration. As the up-regulation of ESM-1 was significant in the presence of the latter, gene silencing experiments with specific siRNA were carried out in 20-mM glucose-derived AGE-HSA-treated HUVECs only. HUVECs were transfected with receptor-specific siRNA and then assayed for ESM-1 expression by ELISA in the presence of AGE-HSA (20-mM glucose derived).
RAGE exerts stimulatory effect on AGE-HSA-induced ESM-1 protein in HUVECs
It was found that both 12-mM (p < 0.005) and 20-mM (p < 0.0005) glucose-derived AGE-HSA caused significant elevation in the expression of RAGE followed by substantial reduction (72%) in protein expression after transfection in the case of 20-mM AGE-HSA (Figure 2(A) and (B)). The expression of ESM-1 was found to be specifically and significantly (19%; p < 0.05) decreased in HUVECs after treatment with RAGE siRNA duplex as compared to mock transfected (no siRNA) cells (Figure 3).
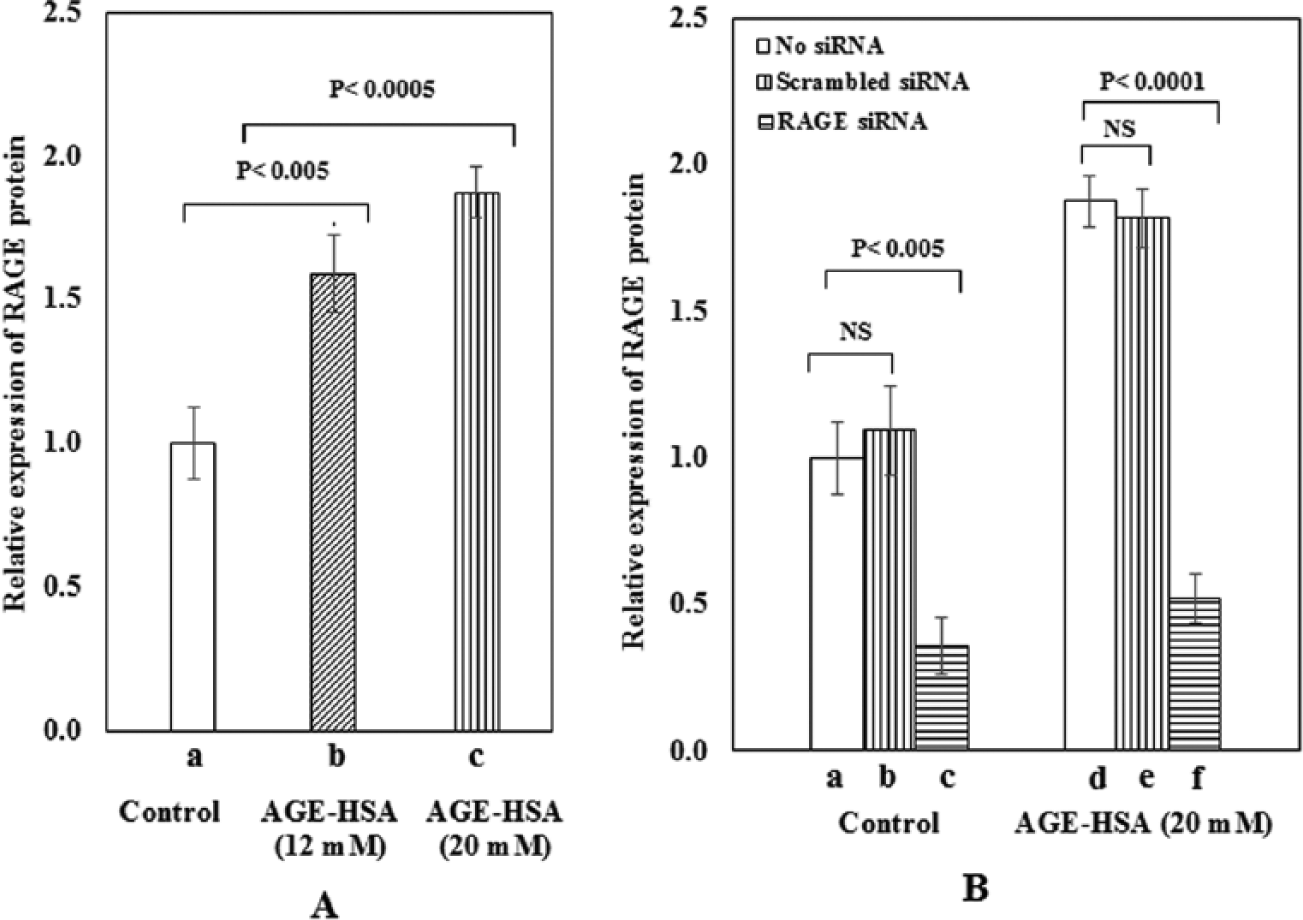
(A) Relative expression of RAGE protein as a function of AGE-HSA: (a) control, (b) 12-mM glucose-derived AGE-HSA (2 mg/mL), and (c) 20-mM glucose-derived AGE-HSA (2 mg/mL). Data are expressed as mean ± standard deviation, n = 3 (paired t-test; p < 0.05 was considered significant). (B) Suppression of RAGE protein expression in the presence of RAGE siRNA. Control (a, b, c; no treatment, in the presence of scrambled siRNA, in the presence of RAGE siRNA, respectively). 20-mM glucose-derived AGE-HSA at 2 mg/mL [d, e, f; AGE-HSA, AGE-HSA along with scrambled siRNA, AGE-HSA along with target siRNA, respectively (n = 3)].
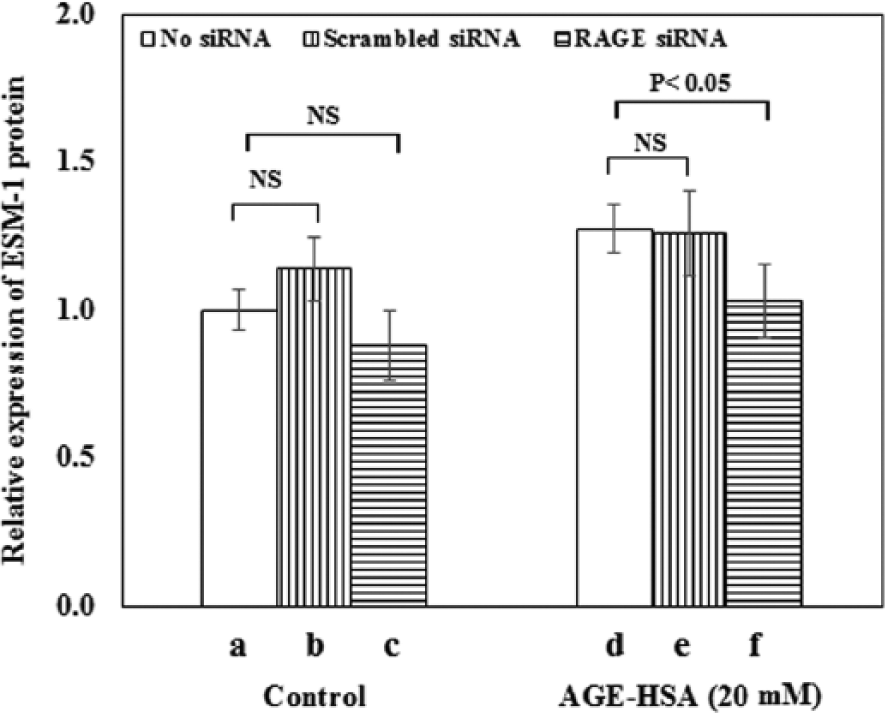
Expression of ESM-1 in HUVEC in response to treatment with RAGE siRNA. HUVECs were subsequently exposed to 20-mM glucose-derived AGE-HSA (2.0 mg/mL). Expression was measured in the absence of siRNA (blank), in the presence of scrambled siRNA (mock) and in the presence of target siRNA. It was reduced in both groups; reduction was significant in AGE-HSA-treated HUVECs only (p < 0.05, n = 3).
Galectin-3 exerts inhibitory effect on AGE-HSA-induced ESM-1 protein in HUVECs
Galectin-3 expression was also significantly up-regulated in both the treated samples as compared to control (12-mM (p < 0.05) and 20-mM glucose-derived AGE-HSA (p < 0.005); Figure 4(A)). Reduction in galectin-3 expression by 76% in the case of 20-mM glucose-derived AGE-HSA confirmed the transfection (Figure 4(B)). The effect of galectin-3 siRNA duplex treatment on ESM-1 expression was found to be opposite to that seen in the case of RAGE siRNA. A significant increase by 22% (p < 0.05) was observed compared with mock transfections (no siRNA) or transfections with a control fluorescein conjugate-scrambled siRNA whose sequence is unrelated to galectin-3 (Figure 5). This suggests the involvement of both AGE receptors in the regulation of ESM-1 expression in HUVEC after treatment with AGE-HSA.
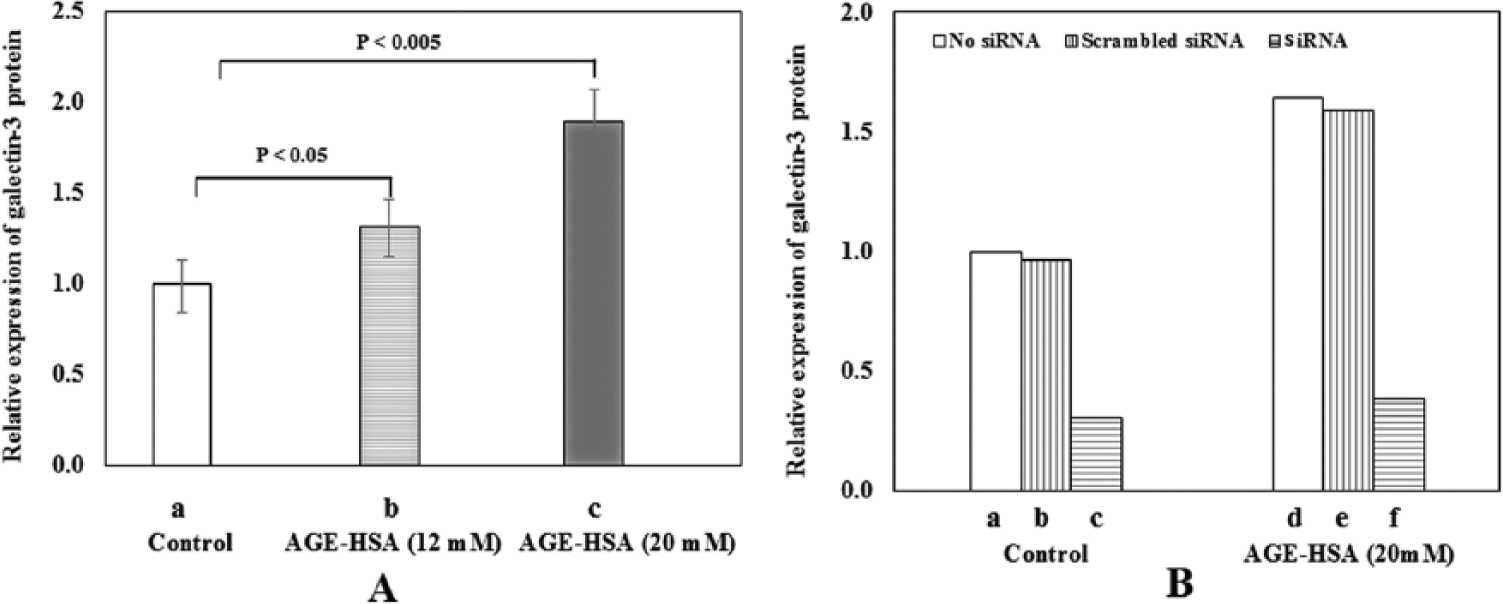
(A) Relative expression of galectin-3 protein as a function of AGE-HSA: (a) control, (b) 12-mM glucose-derived AGE-HSA (2 mg/mL) and (c) 20-mM glucose-derived AGE-HSA (2 mg/mL). Data are expressed as mean ± standard deviation, n = 3 (paired t-test; p < 0.05 was considered significant). (B) Suppression of galectin-3 protein expression in the presence of galectin-3 siRNA. Control (a, b, c; no treatment, in the presence of scrambled siRNA, in the presence of galectin-3 siRNA, respectively). 20-mM glucose-derived AGE-HSA at 2 mg/mL [d, e, f; AGE-HSA, AGE-HSA along with scrambled siRNA, AGE-HSA along with target siRNA, respectively (n = 1)].
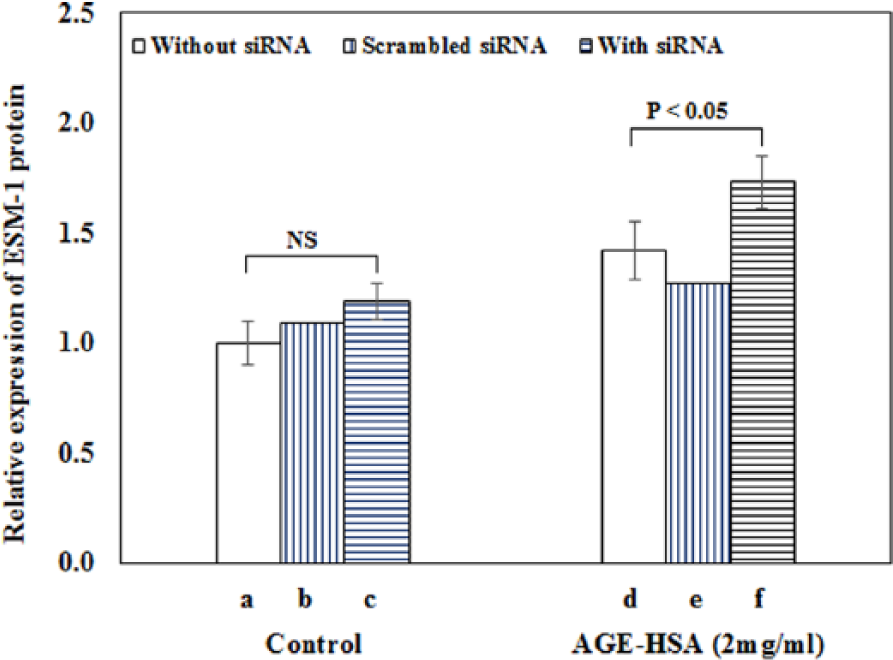
Expression of ESM-1 in HUVEC in response to treatment with galectin-3 siRNA. HUVECs were subsequently exposed to 20-mM glucose-derived AGE-HSA (2.0 mg/mL). Expression was measured in the absence of siRNA (blank), in the presence of scrambled siRNA (mock) and in the presence of target siRNA. It was enhanced in both groups; increase was significant in AGE-HSA-treated HUVECs only (p < 0.05, n = 3).
Regulation of ESM-1 transcription by intracellular signalling pathways
To determine whether inflammatory stress-stimulated MAPK activities had any role in ESM-1 expression, the effects of MAPK inhibitors MAPKKK Activation Inhibitor 570100-5MGCN and JNK inhibitor II 420119-5MGCN on AGE-HSA-stimulated HUVECs were evaluated by quantitative ELISA assay as shown in Figure 6(f) and (g). Expression of ESM-1 was not changed indicating that specific inhibition of ESM-1 synthesis was not mediated by MAPK inhibitors. In contrast, the expression of ESM-1 increased significantly both in controls (p < 0.001) and 20-mM glucose-derived AGE-HSA-treated cells (p < 0.0005; Figure 6(d) and (h)) in the presence of PI3K inhibitor 526559-5MGCN indicating that it exerts a suppressive role. The over-expression demonstrates negative regulation of ESM-1.
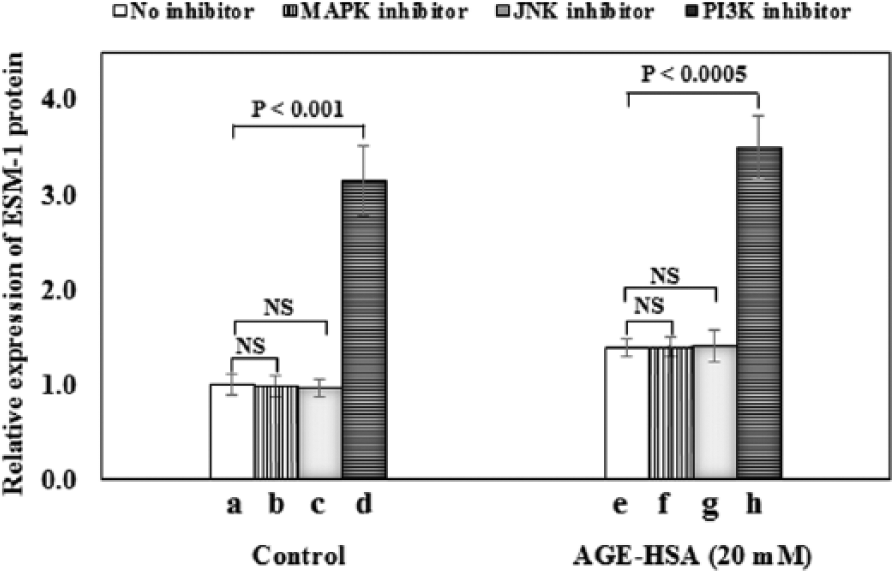
Regulation of AGE-HSA-induced ESM-1 protein expression in HUVECs through different signalling pathway inhibition. Control cells (a, b, c, d; no treatment, 10-µM MAPKKK inhibitor, 10-µM JNK-2 inhibitor and 10-µM PI3K inhibitor, respectively). Cells treated with 20-mM glucose-derived AGE-HSA (e, f, g, h; AGE-HSA, AGE-HSA + 10-µM MAPKKK inhibitor, AGE-HSA + 10-µM JNK-2 inhibitor, and AGE-HSA + 10-µM PI3K inhibitor). Data are expressed as mean ± standard deviation, n = 3 (paired t-test; p < 0.05 was considered significant).
Discussion
We report here that glycated HSA stimulates production of ESM-1 in primary HUVECs. ESM-1 was first reported to be a tumour endothelial cell specific marker and a potential key player in cancer and inflammation. In sepsis, it has been again shown to be an indicator of vascular health. Stimulation of vascular smooth muscle cell proliferation and migration by ESM-1 during atherogenesis has also been observed. 18 In another study, ESM-1 was not detected in quiescent tissues or endothelium of great arteries. 19 Overall, it is now established as a potential biomarker for endothelial activation or dysfunction. Even though it is a freely circulating proteoglycan, no direct evidence of ESM-1 in blood samples of diabetic patients has been reported so far, and its role in diabetes remains unknown. Our results demonstrate a distinct in vitro expression of ESM-1 in HUVECs, which is modulated by glycated protein in a dose-dependent manner. The only other study we came across reports elevated vitreous endocan levels in relation to PDR in patients compared to non-diabetic individuals. 13 The serum levels of endocan were, however, similar in diabetic patients and non-diabetic individuals. We have made a similar observation in serum ESM-1 levels taken from a cross-section of diabetic individuals and healthy volunteers (unpublished observations). Our previous studies undertaken with AGE-HSA at these concentrations have confirmed activation of HUVECs through up-regulation of the inflammatory marker sICAM-1 and impairment of NO synthase activity. 20
In order to confirm up-regulation of ESM-1 as a result of AGE-induced inflammatory stress, we have investigated the role of two receptors of AGEs, namely, RAGE and galectin-3 receptors. First, we demonstrate the secretion of galectin-3 in AGE-activated HUVEC which is an endothelial cell type. We observe that galectin-3 AGE receptor is expressed in HUVECs in quiescent state and when challenged with AGE-HSA in a dose-dependent manner. This is in agreement with previous studies about target tissues of diabetic vascular pathology such as endothelium and mesangium, which report weak expression of galectin-3 under basal conditions but induction of its expression when exposed to high glucose concentrations or AGEs.21,22
In HUVECs transfected with RAGE siRNA, AGE-HSA-induced ESM-1 expression was decreased by 19% as compared to non-transfected cells, indicating a direct role of AGE–RAGE binding in ESM-1 expression. AGE–RAGE interaction activates several inflammatory signalling pathways, possibly leading to its up-regulation. Due to the RAGE gene silencing, the activation of inflammatory signalling pathways was blocked, and as a result, decrease in ESM-1 expression was observed. In the case of HUVECs transfected with galectin-3 siRNA, ESM-1 expression was increased by 22% in the presence of AGE-HSA as compared to non-transfected cells, indicating indirectly AGE–galectin-3 binding and hence reduced activity of AGE-HSA. Galectin-3 has not been shown to transduce cellular signals after binding with AGEs but is considered to function in a protective manner in vivo, as a sequestering AGE receptor, towards AGE-dependent tissue injury.23,24 The mechanisms by which AGEs induce ESM-1 are yet to be elucidated, but it appears that the AGE receptors and their signalling cascade may play a role in its regulation, since silencing of RAGE gene causes down-regulation, whereas the silencing of galectin-3 gene causes up-regulation in the ESM-1 expression. The inflammatory signalling pathways activated by AGE–RAGE binding, especially the activation of VEGF, may be involved in the up-regulation of ESM-1 protein expression, reported earlier. 25 Due to the RAGE gene silencing, these inflammatory signalling pathways get suppressed as a result of which down-regulation in the expression of ESM-1 protein was observed. Galectin-3, on the other hand, does not activate AGE-induced inflammation unlike RAGE, since it lacks the signalling sequence that triggers inflammatory responses. So the silencing of galectin-3 gene shows up-regulation in ESM-1 expression. Thus, the two receptors function in an antagonistic manner. Additionally, when galectin-3 concentration increases, the chances of AGE–RAGE interaction decrease because a competition between RAGE and galectin-3 for binding with the AGE ligands is likely to be initiated. This may also be a reason for the enhancement of ESM-1 expression when galectin-3 was silenced by siRNA. The action of galectin-3 receptor appears to vary with the cell type as shown in in vivo studies conducted on galectin-3 knockout mice. 26 The galectin-3 deficient mice fed on modified lipid-rich diet developed inflammatory lesions in the aortic wall vis-à-vis their wild type counterparts where only fatty streaks were seen. Recruitment of activated T lymphocytes and marked monocyte-macrophage infiltration accompanied by inflammatory mediator expression was also observed. Additionally, up-regulation of the pro-inflammatory receptor RAGE was noticed. These findings strongly suggest the protective role played by galectin-3 in the vascular tissue via uptake and effective removal of oxidized lipoproteins leading to down-regulation of pro-inflammatory pathways associated with atherogenesis. In the case of nervous tissue, it has been shown through in vitro studies in microglial cells 27 and in vivo experiments in galectin-3 knockout mice that galectin-3 is associated with neuronal regeneration. 28
The mechanisms by which AGEs induce ESM-1 are yet to be elucidated. Mechanistic investigations indicated that inhibition of MAPK pathway did not influence basal or AGE-HSA-induced ESM-1 expression indicating that MAPK signalling has no modulatory role. Inhibition of JNK signalling cascade which mediates inflammatory events also did not affect the ability of AGE-HSA to induce ESM-1 expression significantly. However, inhibition of PI3K caused a threefold increase suggesting a suppressive function of PI3K. Inactivation of PI3K caused increase in ESM-1 expression that was higher by 150% as compared to AGE-HSA-induced response. Involvement of PI3K pathway has been shown earlier in human renal cancer cells. 25 Previous studies in tumour cells and HUVECs have demonstrated a positive correlation between VEGF levels and endocan mRNA levels.29,30 Investigations in cultured blood vascular endothelial cells have revealed that endocan expression was stimulated by vascular endothelial growth factor A (VEGF-A) through the phosphorylation of its receptor. 31 We have also observed in an earlier study that AGE-HSA at the same concentration (2.0 mg/mL) up-regulates VEGF expression in HUVECs. Therefore, we can infer that the AGE-induced expression of ESM-1 in this study is accompanied by VEGF production also. 32
The physiological importance of ESM-1 (endocan) is imparted by its ability to bind growth factors and other molecules due to the hybrid nature of its structural components [chondroitin sulfate/dermatan sulfate (CS/DS)]. Endocan itself has been shown to elicit severe inflammatory responses both in vitro in HUVECs and in vivo in mice model. 33 Joladarashi et al. 34 have reported structural changes in certain moieties of proteoglycans in diabetic rat kidney cells which may lead to increased interactions of these molecules with extracellular matrix (ECM). This may account for the absence of detectable increase in serum ESM-1 levels in diabetic patients, whereas a marked increase is observed in in vitro experiments, where the lack of ECM precludes any such interaction.
Footnotes
Declaration of conflicting interests
The authors declare that they have no conflicts of interest.
Funding
This work has been supported by institutional grants. Bikesh K Nirala was supported by a student fellowship from University Grants Commission, India.