
Abstract
Traditional treatments for type 1 and type 2 diabetes are often associated with side effects, including weight gain and hypoglycaemia that may offset the benefits of blood glucose lowering. The kidneys filter and reabsorb large amounts of glucose, and urine is almost free of glucose in normoglycaemia. The sodium-dependent glucose transporter (SGLT)-2 in the early proximal tubule reabsorbs the majority of filtered glucose. Remaining glucose is reabsorbed by SGLT1 in the late proximal tubule. Diabetes enhances renal glucose reabsorption by increasing the tubular glucose load and the expression of SGLT2 (as shown in mice), which maintains hyperglycaemia. Inhibitors of SGLT2 enhance urinary glucose excretion and thereby lower blood glucose levels in type 1 and type 2 diabetes. The load-dependent increase in SGLT1-mediated glucose reabsorption explains why SGLT2 inhibitors in normoglycaemic conditions enhance urinary glucose excretion to only ~50% of the filtered glucose. The role of SGLT1 in both renal and intestinal glucose reabsorption provides a rationale for the development of dual SGLT1/2 inhibitors. SGLT2 inhibitors lower blood glucose levels independent of insulin and induce pleiotropic actions that may be relevant in the context of lowering cardiovascular risk. Ongoing long-term clinical studies will determine whether SGLT2 inhibitors have a safety profile and exert cardiovascular benefits that are superior to traditional agents.
Introduction
Diabetes mellitus affects more than 380 million individuals worldwide. 1 It is the leading cause of end-stage kidney failure, and after accounting for traditional risk factors such as obesity and hypertension, diabetes remains a significant (approximately twofold) risk factor for cardiovascular disease. 2 Indeed, the majority of individuals with diabetes will die from cardiovascular disease. Complications are generally considered to arise from chronic hyperglycaemia, and a primary aim in the management of diabetes is to improve glycaemic control (HbA1c < 7%).
Numerous trials have been carried out to ascertain the benefits of intensive glucose lowering on cardiovascular outcomes in type 2 diabetes.3–6 Despite reductions in blood glucose to almost normal levels, using traditional therapies such as metformin, sulphonylureas and insulin, mortality from cardiovascular disease was either increased 5 or not affected.3,4,6 On the other hand, a post-trial follow-up of one of these studies revealed that intensive glucose control reduced the risk of microvascular disease, myocardial infarction and death from any cause at 10 years despite increasing blood glucose levels within 1 year after the trial. 7 Similarly, in type 1 diabetes, intensive therapy for greater than 6 years reduced the number of cardiovascular events in the long term. 8 The few definitive studies on cardiovascular benefits in diabetes have somewhat dampened the urgency for intensive glucose control in the clinic.
Traditional anti-diabetic agents come with a number of side effects that may outweigh the benefits from blood glucose lowering and/or compromise patient compliance. Metformin is the first line therapy for type 2 diabetes and lowers fasting blood glucose levels via inhibitory effects on hepatic and renal gluconeogenesis. However, gastrointestinal upset is a common side effect. 9 Similarly, targeting the incretin hormone system with glucagon-like peptide-1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP-4) inhibitors can be associated with nausea and vomiting, and peroxisome proliferator-activated receptor (PPAR) agonists (glitazones) are linked with oedema and weight gain. 9 There is also evidence to suggest that insulin secretagogues, such as sulphonylureas, exacerbate myocardial ischaemia, infarct area and heart failure. 10 The glucose lowering effects of many anti-diabetic compounds presently available are dependent on endogenous insulin. However, in type 2 diabetes, progressive β-cell failure is a major barrier for these agents to achieve their intended effects, such that the use of multiple agents including insulin is eventually required. Exogenous insulin and insulin secretagogues significantly increase the risk of hypoglycaemia and weight gain, which are independent risk factors for cardiovascular disease. 11 Therefore, treatment strategies that effectively reduce hyperglycaemia, in the absence of counteractive side effects, are in great need.
Sodium-dependent glucose transporter (SGLT)-2 inhibitors have recently entered the market as a promising anti-diabetic strategy (for review, see Hasan et al. 12 ). This class of medication targets the kidney proximal tubules to block glucose reabsorption, thereby enhancing urinary glucose excretion and conferring anti-hyperglycaemic effects that are primarily independent of insulin. Here, we review the physiology of glucose reabsorption by the kidney, the basic biology of SGLTs and consider the implications of targeting SGLT as a new therapeutic approach for the management of blood glucose levels in diabetes.
Glucose handling by the kidney
Plasma glucose is usually maintained within the range of 3.9–8.9 mmol/L due to an elaborate control system involving intestinal absorption, glucose production, renal reabsorption, utilization and excretion by a number of tissues in the body. This is essential to ensure the brain receives a constant source of glucose, which is its major fuel for energy production and adequate function. The kidney is a site of significant glucose handling, which includes glomerular filtration and proximal tubule reabsorption of glucose, as well as contributing to endogenous glucose production (EGP; gluconeogenesis). Renal gluconeogenesis, which is primarily localized in the proximal tubule, is especially important for glucose homeostasis during times of fasting and is upregulated in diabetes., In type 2 diabetes, the kidney contributes equally with hepatic sources to enhanced gluconeogenesis. 13
Glucose reabsorption by the kidney
Under conditions of normal blood glucose levels and normal glomerular filtration rate (GFR), the kidneys filter 160–180 g of glucose each day [~5.5 mmol/L (100 mg/dL) × 180 L/day]. This would result in a urinary loss of energy substrate equal to ~30% of the daily energy expenditure if not regained by the renal tubules. Renal glucose reabsorption occurs primarily in the proximal tubules of the kidney. In a healthy individual, the maximum transport capacity (Tmax) of the proximal tubules is ~500 g of glucose/day, and therefore, virtually all of the filtered glucose is normally reabsorbed by the kidneys. Theoretically, this Tmax would result in a plasma glucose threshold of ~15.5 mmol/L before any glucose becomes present in the urine. However, due to variability in the Tmax for individual nephrons, the observed plasma glucose concentration that results in glucosuria in a normal glucose-tolerant individual is ~10–11.1 mmol/L. It is worth noting that proximal tubule cells do not utilize glucose to a significant extent for the production of energy, but glucose is primarily returned to the circulation. 14 As a consequence, much research is ongoing to determine whether targeting renal glucose reabsorption, that is, an ‘insulin-independent’ pathway, can provide benefits in diabetes that are superior to traditional approaches.
Membrane-associated transport proteins are responsible for reabsorbing glucose, a polar molecule, across the luminal and basolateral plasma membrane of proximal tubular cells: SGLT1 and SGLT2 are secondary active co-transporters located on the apical or luminal membrane (Figure 1). The sodium potassium adenosine triphosphatase active transporter (Na+/K+ ATPase) is located on the basolateral membrane and establishes the concentration gradient that drives Na+, and thereby glucose, via SGLT1 and SGLT2 into the cell from the luminal surface. The facilitative glucose transporter (GLUT2) is found on the basolateral membrane and is responsible for the majority of concentration gradient–driven exit of glucose from the cells into the interstitium and peritubular circulation.
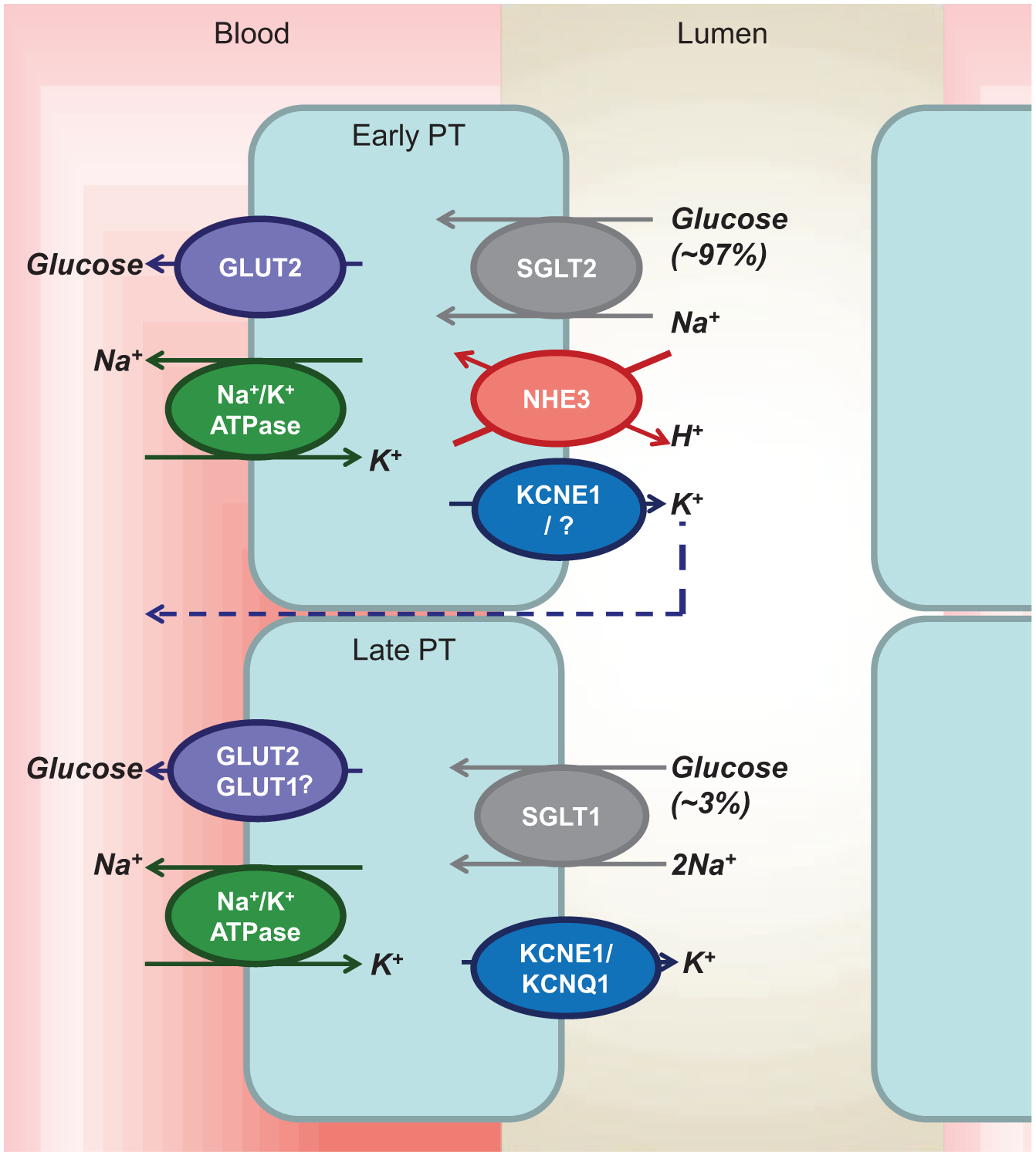
Glucose reabsorption in the renal proximal tubule. The basolateral Na+/K+ ATPase pumps Na+ out and K+ into the cell to establish an inward Na+ gradient. This gradient is used for Na+ and glucose co-transport across the luminal brush border of the early proximal tubule through SGLT2, and the glucose is passively returned via GLUT2 to the interstitium/bloodstream. In the late proximal tubule, SGLT1 is responsible for ‘mopping up’ remaining luminal glucose, while the role of basolateral GLUT1-facilitated glucose transport in this segment remains unclear. Apical efflux of K+ maintains the electrogenic gradient.
Biology of the SGLT1 and SGLT2
Experiments performed on isolated nephron segments of rabbit kidneys in the early 1980s identified differences between the early and late proximal tubule segments, with respect to the rate of uptake and affinity for glucose. 15 Later studies confirmed that the heterogeneity in Na+-glucose transport across the proximal tubule was attributed to the presence of two different glucose transporters along the apical surface. 16
SGLT1 and SGLT2 have been the most intensively studied of the human solute carrier family 5 (SLC5), which now includes 12 members. Six of these are named as SGLTs, varying in their preferences for sugar binding (Table 1). Others in the SLC5 family include sodium co-transporters for myo-inositol (SMIT1), iodide (NIS), monocarboxylic acid (SMCT), multivitamin (SMVT) or choline (CHT). 18 The molecular nature of SGLTs has been largely pioneered by studies in the laboratory of Wright and colleagues,19,20 which involved identifying and cloning the SGLT1, identifying that defects in SGLT1 were associated with intestinal malabsorption of glucose–galactose, 21 cloning SGLT2 22 and defining the crystal structure of a sodium galactose bacterial isoform in Vibrio parahaemolyticus (vSGLT), 23 which subsequently allowed for the discovery of how Na+ and sugar transport is coupled. 24 The reader is referred to a comprehensive review for a detailed history on the research efforts that led to our current understanding of the SGLTs. 17
Natural substrates of the six SGLTs in the human body.
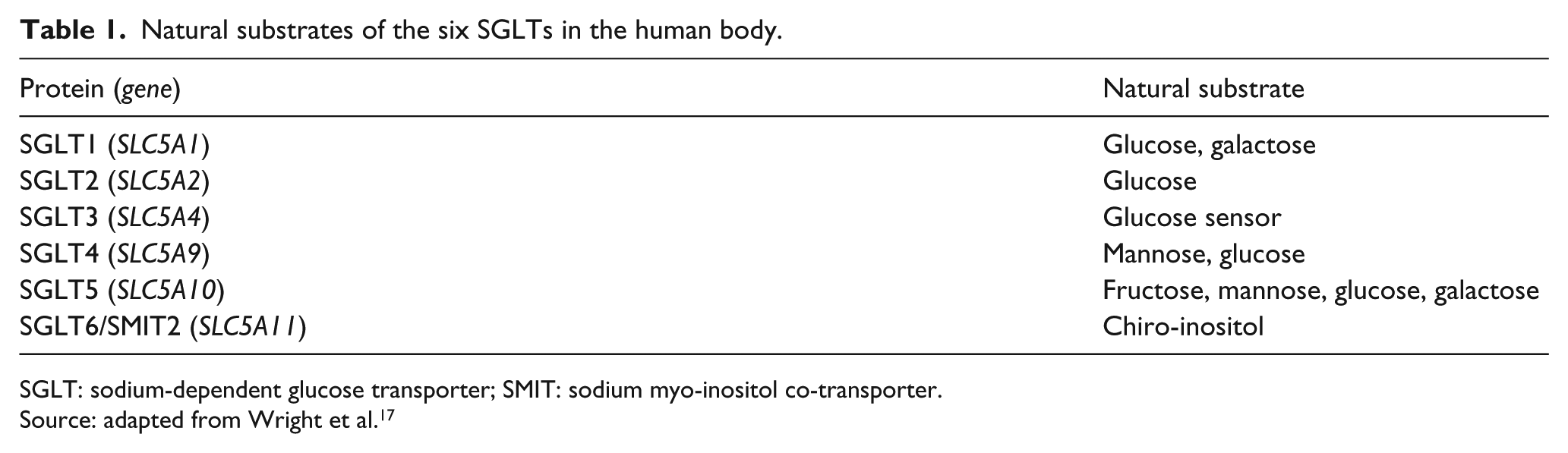
SGLT: sodium-dependent glucose transporter; SMIT: sodium myo-inositol co-transporter.
Source: adapted from Wright et al. 17
All SGLTs have 15 exons, spanning from 8 to 72 kb which code for 60- to 80-kDa proteins that comprised 580–718 amino acids.
17
Human SGLT1 (hSGLT1) consists of 14 transmembrane α-helices with the hydrophobic NH2 and COOH termini facing the extracellular space. Electrophysiological techniques have been used to study sugar selectivity and transport kinetics of cloned SGLTs in a range of expression systems.
17
Using transfected human embryonic kidney (HEK) 293T cells, the apparent affinities (Km) for
In the early 1980s, Peerce and Wright 19 and Schmidt et al. 30 described the existence of SGLT1 as a 73-kDa protein through a series of experiments using azido-phlorizin-photoaffinity labelling and antibodies. Determining the distribution of SGLT proteins has been somewhat hampered due to the lack of specific antibodies. However, with the use of well-validated antibodies, SGLT1 has been localized to the brush border membrane of the small intestine and late proximal tubule31–35 and SGLT2 to the brush border membrane of the early proximal tubule. 27 Using positron emission tomography (PET), the location of active transporters can be safely determined in human subjects. 17 α-methyl-4-deoxy-4-[ 18 F]fluoro-glucopyranoside (α-Me4FDG) is a Na+-dependent non-metabolizable substrate that has a high affinity for SGLT1 and SGLT2 but not GLUTs. Me4FDG was distributed within the kidney, skeletal muscle, heart, liver, prostate, uterus and testes. The lack of Me4FDG accumulation in the bladder was consistent with complete renal tubular reabsorption by SGLTs. Ongoing studies will serve to establish which SGLTs are responsible for the uptake of Me4FDG in these extrarenal tissues across a range of species.
Individuals carrying mutations have provided evidence for the relative contribution of SGLT1 and SGLT2 to renal glucose reabsorption. Mutations in the SLC5A2 gene (encoding SGLT2) are associated with intact intestinal glucose reabsorption but persistent glucosuria (ranging from 1 to >100 g/day; familial renal glucosuria), whereas mutations in SLC5A1 (encoding SGLT1) result in intestinal malabsorption of glucose and galactose but negligible glucosuria.36,37 Using free-flow micropuncture, it has been demonstrated that glucose reabsorption in the early proximal tubule is completely absent in Sglt2−/− mice, and that SGLT2 is, in fact, responsible for the bulk (>90%) of glucose reabsorption by the kidney in normoglycaemia. 27 Gene knockout of SGLT1 in mice induced a small increase in fractional glucose excretion to ~3%; 38 these results are consistent with the maximum phenotypes described for individuals with mutations in the genes encoding these transporters. 39 Net renal glucose reabsorption is absent in Sglt1/Sglt2−/− mice, which confirms that these two transporters account for all glucose reabsorption in the kidney under normoglycaemic conditions. 40
Glucose reabsorption in the diabetic kidney
In diabetes, the filtered glucose load increases due to increased blood glucose concentrations and, in some instances, increased GFR. For example, a blood glucose concentration of 10–12 mmol/L (and normal GFR) would result in ~324–390 g/day of filtered glucose. Despite this, there can be very little increase in urinary glucose excretion as the Tmax for glucose can increase from ~500 g/day in a healthy individual to 600 g/day in type 1 and type 2 diabetes.41,42 If the filtered glucose load exceeds the Tmax of proximal tubules, glucosuria increases in a linear fashion. Proximal tubule growth is a key feature in the early stages of diabetes that may explain the increased capacity for renal glucose reabsorption. Tubule growth involves a unique molecular signature including a period of hyperplasia followed by hypertrophy and aspects of senescence, which also may set the stage for inflammation, fibrosis and diabetic nephropathy. 43
More specifically, changes in activity and expression levels of glucose transporters may contribute to the enhanced renal Tmax for glucose in diabetes. While there is currently no convincing evidence on how SGLT2 expression changes in diabetic humans, animal studies have demonstrated that SGLT2 protein expression (using a renal membrane preparation) was increased by almost 2-fold in the whole kidney of Ins2+/C96Y Akita and db/db mouse models of type 1 and type 2 diabetes, respectively, and knockout mice were used as a negative control for the antibody. 44 In contrast, streptozotocin (STZ)-induced diabetic mice had decreased renal SGLT2 expression, 44 which is possibly a direct effect of STZ on the proximal tubule. These data suggest that the early proximal tubule is further increasing its capacity to reabsorb glucose in diabetes, which will not only exacerbate hyperglycaemia but theoretically could enhance glycotoxicity in proximal tubular cells.45,46 SGLT1 protein expression, on the other hand, becomes suppressed when the late proximal tubule is exposed to increased glucose, as evident when SGLT2 is lacking or inhibited and when SGLT2s become saturated in diabetes.27,47 This may be an attempt to protect the late proximal tubule from excessive glucose exposure, albeit the molecular mechanisms for this differential response to glucose by the two transporters are unknown.
Interestingly, the application of insulin to HEK-293T cells expressing hSGLT2 increased Na+-glucose transport, which was completely dependent on phosphorylation of the ser624 residue. 48 This is perhaps not surprising given that insulin binding coincides greatly with the location of SGLT2 in the proximal tubule 49 and that insulin release following a meal may act on the proximal tubule to conserve filtered glucose. Moreover, hyperinsulinemia in the db/db mouse might contribute to the increased membrane expression levels of SGLT2 in this model of type 2 diabetes.44,50 However, the Akita mouse model of type 1 diabetes has low insulin levels but similarly increased renal membrane SGLT2 protein expression,44,47 indicating that regulators other than insulin are also at play (e.g. tubular growth, see above). With regard to SGLT1, insulin stimulation slightly decreased Na+-glucose transport in HEK-293T cells, 48 further highlighting a disparity in the regulation of these two transporters. In addition, SGLT1 messenger RNA (mRNA) and protein levels did not change in parallel in diabetic mice. 47 Any variability in anti-hyperglycaemic and pleiotropic effects (see below) upon SGLT2 inhibition may be explained, in part, by differences in the expression and activity levels of SGLT1/2. A better understanding of the molecular signals that determine transporter expression and activity may help to further refine therapies in the kidney to lower blood glucose levels.
SGLT2 and SGLT1 as therapeutic targets
In 1835, phlorizin was isolated from the bark of apple trees by French chemists 51 and was subsequently demonstrated to promote glucosuria and lower serum glucose levels in a diabetic patient 52 and completely inhibit the renal reabsorption of glucose in humans. 53 It was not pursued for clinical use due to poor intestinal absorption, low bioavailability and lack of selectivity for SGLT2. Additionally, phlorizin is hydrolyzed to phloretin in the gut, which has been shown to inhibit multiple GLUTs.17,54 Efforts to develop a derivative of this compound were not renewed until the late 1980s when Rossetti et al. 55 identified that phlorizin normalized insulin sensitivity and plasma glucose levels and that these effects were completely reversed when the drug was withdrawn. The subsequent cloning of hSGLTs20,22 provided further impetus, and the first phlorizin derivative, T-1095, was developed. This compound was also non-selective. Sergliflozin and remogliflozin were subsequently developed, with greater affinities for SGLT2, and the former entered clinical trials. The major problem with many of the early agents was that O-glucoside linkages made them susceptible to hydrolysis by β-glucosidases and the lower plasma half-lives rendered them unsuitable for clinical use. Since then, much progress has been made with regard to developing molecules that are more selective for SGLT2, and due to their C-glucoside configuration, these newer drugs do not undergo enzyme-mediated hydrolysis. This has resulted in the development of long-acting SGLT2 inhibitors for use as a once-daily oral treatment in diabetes.
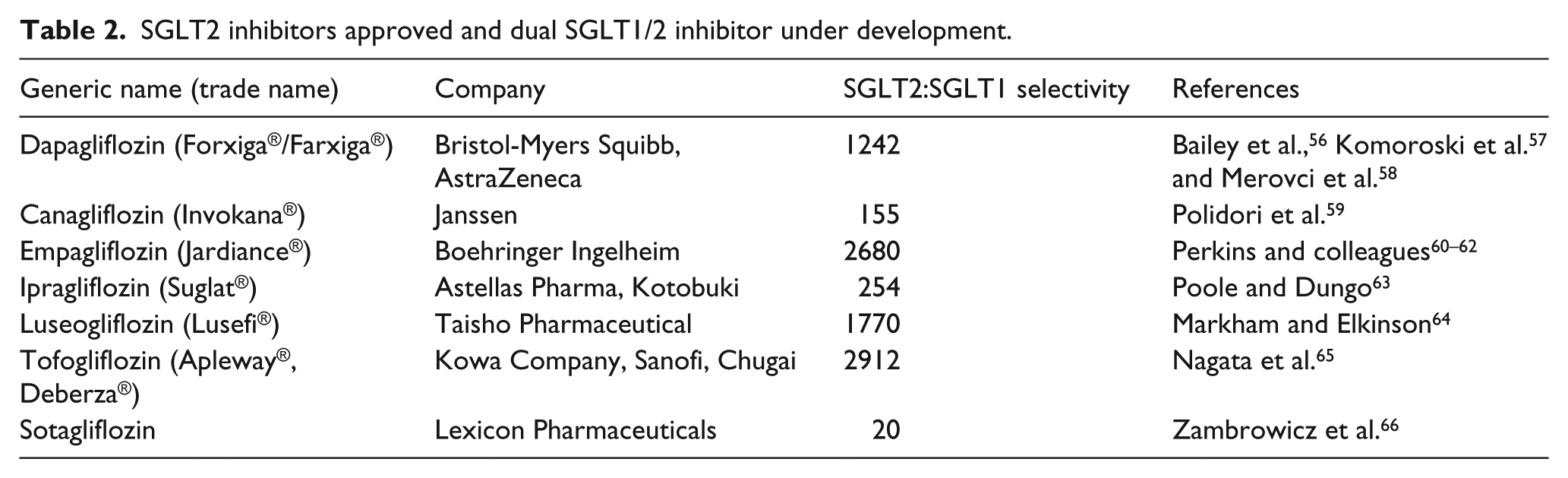
A number of SGLT2 inhibitors have been developed and tested in preclinical models through to post-marketing surveillance (Table 2). Three members in this class, dapagliflozin (Forxiga® or Farxiga® in the United States), canagliflozin (Invokana®) and empagliflozin (Jardiance®) are approved in the United States and Europe, and the two former members are also available in Australia. Others including ipragliflozin (Suglat®), luseogliflozin (Lusefi®) and tofogliflozin (Apleway®, Deberza®) are approved in Japan. They are indicated for use in type 2 diabetes without kidney disease and are under clinical investigation as an add-on to insulin in type 1 diabetes. Clinical studies have reported significant reductions in fasting plasma glucose and HbA1c levels (0.7%–0.8%) with SGLT2 inhibitors compared to placebo and active comparators.56,60–62,67 Their mechanism of action is independent of insulin, and therefore, they are thought to be effective at any stage of progressive β-cell failure and/or insulin resistance, given that the kidneys are functioning adequately. Moreover, when used alone or in conjunction with other traditional agents, this novel therapy confers very little hypoglycaemic risk.
SGLT2 inhibitors approved and dual SGLT1/2 inhibitor under development.
SGLT2 inhibition unmasks the transport capacity of SGLT1
Given that SGLT2 is responsible for >90% of glucose reabsorption by the kidney, its inhibition would be expected to induce a urinary glucose loss that is close to the filtered load (160–180 g/day in normoglycaemia). However, while all SGLT2 inhibitors produce a dose-dependent glucosuria, they often induce a maximum urinary glucose loss of ~40–80 g/day in healthy individuals 57 and in type 2 diabetes, 57 indicating that significant glucose reabsorption persists. This is despite complete inhibition of SGLT2 in vitro at nanomolar concentrations.68,69 An explanation for the maintenance of significant renal glucose reabsorption upon SGLT2 inhibition is the apparent reserve capacity of SGLT1. Maximal dosing with an SGLT2 inhibitor in a normal glucose-tolerant individual should theoretically provide an estimate of the Tmax for SGLT1, assuming the tubular concentrations were high enough to effectively compete with the increased luminal glucose concentrations following SGLT2 inhibition. Individuals with mutations in the gene encoding SGLT2 (familial glucosuria) excrete, in some cases, >100 g of glucose/day. 39 While this indicates that the Tmax for SGLT1 may be close to 80 g/day, the quantitative effect of these genetic mutations on total SGLT2 activity has not been determined. As an alternative approach, genetic mouse models have allowed for more accurate determination of SGLT1-mediated glucose reabsorption in the absence of SGLT2. In Sglt2−/− mice, glucose reabsorption in the early proximal tubule was completely absent, but fractional glucose reabsorption at the level of the whole kidney was maintained at an average of 36% under normoglycaemia, 27 which is close to the values seen in healthy human subjects administered with an SGLT2 inhibitor. 57 This was associated with greatly increased glucose reabsorption in the late proximal tubule, 27 coinciding with the site of SGLT1. Subsequent studies in Sglt2−/− mice showed that the additional knockout of Sglt1 significantly increased glucosuria. 70 Further work by the Vallon laboratory then exactly quantified glucose handling in renal clearance studies and identified that net renal glucose reabsorption was completely absent in Sglt1−/− mice treated with the SGLT2 inhibitor, empagliflozin, and in Sglt1/Sglt2−/− mice. 40 This confirmed that increased SGLT1-mediated glucose uptake fully explains the maintenance of significant glucose reabsorption when SGLT2 is inhibited in normoglycaemia (and likely in cases of familial glucosuria where SGLT2 is absent, that is, those with premature stop codon mutations). Yet to be directly tested is whether SGLT2 and SGLT1 account for all renal glucose reabsorption in diabetes or whether other luminal glucose transporters contribute.
Rationale for dual SGLT1/2 inhibition
The mouse studies described above provided evidence that SGLT2 inhibition causes glucosuria only once the Tmax for SGLT1 has been exceeded (since SGLT1 is downstream of SGLT2 in the proximal tubule). 40 A concern related to inhibition of the SGLT1 is the prospect for gastrointestinal symptoms to arise (i.e. nausea, vomiting, diarrhoea and glucose–galactose malabsorption). 38 As such, pharmaceutical companies have primarily focused on the development of highly selective SGLT2 inhibitors. However, there is now emerging evidence that additional partial inhibition of SGLT1, using canagliflozin (IC50: 155:1) or LX4211/sotagliflozin (IC50: 20:1), may provide additional benefits on glucose homeostasis in the absence of reported gastrointestinal side effects compared with placebo.59,66 Partial inhibition of SGLT1 increases urinary glucose excretion via actions on the kidney and also delays intestinal glucose reabsorption. Increased glucose delivery to the distal gut stimulates post-prandial secretion of GLP-1 and peptide YY.59,66 Therefore, dual SGLT1/2 inhibition may contribute to additional benefits on blood glucose lowering via combined actions on the kidney and gut (see also article by La Puerta et al., this issue). Phase II trials involving sotagliflozin in type 2 diabetes with renal impairment and in type 1 diabetes have been completed and awaiting results (ClinicalTrials.gov Identifier: NCT01742208, NCT01555008).
How do SGLT2 inhibitors reach their target?
Using whole-cell patch clamp recordings, studies by Ghezzi et al. 68 indicated that SGLT2 inhibitors exert their inhibitory effects from the extracellular surface of the tubule lumen and do not function intracellularly. Whether these drugs, in addition to being filtered at the glomerulus, are also secreted or reabsorbed by the renal tubules is not known. In mice treated with empagliflozin, the free plasma concentration and thus the early proximal tubule concentrations of the drug derived from filtration matched ~10- to 20-fold of the IC50 for SGLT2; this was associated with a reduction of fractional glucose reabsorption to ~45% (the remainder being attributed to SGLT1 transport as fractional glucose reabsorption was absent in empagliflozin-treated Sglt1−/− mice). 40 This is close to the mean fractional glucose reabsorption observed in normoglycaemic Sglt2−/− mice. 27 These studies are consistent with the notion that SGLT2 inhibitors are reaching their target, in the brush border membrane of the proximal tubule via glomerular filtration, although the exact role of tubular secretion and/or reabsorption remains to be determined.
Effects on the kidney and beyond
The effects of SGLT2 inhibition on the kidney and subsequent systemic changes are depicted in Figure 2. In type 2 diabetes, treatment with dapagliflozin for 2 weeks increased EGP that was not explained by a reduction in plasma glucose levels. 58 Parallel increases in plasma glucagon levels implicated hepatic sources as the primary contributor to EGP with SGLT2 inhibition; however, the contribution of renal gluconeogenesis was not clear. In the Akita mouse model of type 1 diabetes, empagliflozin attenuated the diabetes-induced overexpression of whole-kidney phosphoenolpyruvate carboxykinase (PEPCK) mRNA, which is a principal gluconeogenic enzyme. 47 This suggests that renal gluconeogenesis may not contribute significantly to enhanced EGP with SGLT2 inhibition, although this remains to be directly tested. Indeed, SGLT2 inhibition establishes a proximal tubule gradient, which may promote an inward flux of glucose via the bidirectional GLUT2 transporter from the basolateral side; it is not known what the fate of glucose may be if this should occur. It also remains unclear how tubular structures, downstream of the early proximal tubule, are affected by increased luminal glucose load in the context of alterations to protein function (i.e. membrane-bound transporters and enzymes). For instance, extracellular glucose has been shown to facilitate GLUT9-mediated exchange for intracellular urate in vitro. 71 Indeed, diabetic individuals with glucosuria had 4% higher fractional excretion of uric acid compared to those without glucosuria, 72 and SGLT2 inhibition with dapagliflozin in type 2 diabetes reduces serum uric acid levels. 73 A direct relationship between luminal glucose and uric acid excretion and whether this contributes to the lowering of hyperuricemia with SGLT2 inhibition in diabetes remain to be tested. Moreover, the effects of glucose entry via GLUT9, which has been primarily localized to the proximal tubule in humans, are unknown. 74 Tubulotoxicity may result from increased levels of glucose that induce inflammatory and pro-fibrotic effects, and the formation of advanced glycation end-products; all of which have been linked to the development and progression of diabetic nephropathy (for review, see Forbes and Cooper 75 ). In non-diabetic mice, the increase in glucose delivery downstream of the early proximal tubule, due to genetic or pharmacologic inhibition of SGLT2, did not alter the expression of kidney injury markers although there were minor increases in markers of tubular growth and protective mechanisms, respectively.44,47,76 It is worth noting the case of one patient with familial glucosuria who had severe urinary glucose excretion (>109 g/day) with no kidney dysfunction at 20 years after diagnosis. 77 However, as noted above, it is a rare condition and not studied in detail.
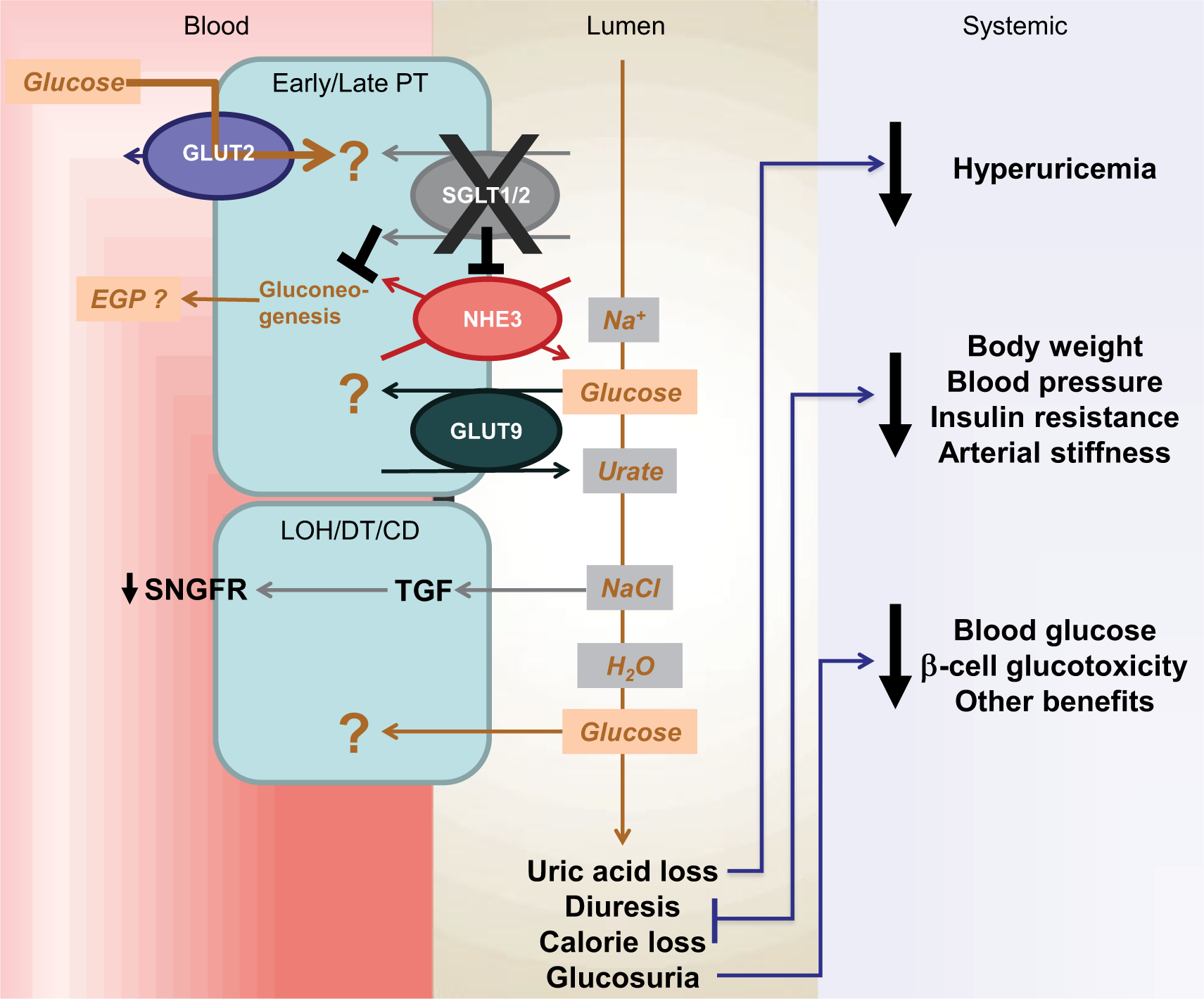
Downstream effects of proximal tubule SGLT1/2 inhibition. Glucose is freely filtered at the glomerulus into the tubular lumen, and normally, all of it is reabsorbed along the apical brush border membrane by the SGLT2 (and SGLT1 to a lesser extent) in the proximal tubule. Inhibition of SGLT2 or dual inhibition of SGLT1/2 establishes a concentration gradient that may drive glucose entry from the peritubular capillaries via the bidirectional GLUT2 transporter. The fate of glucose entering proximal tubule cells via this pathway is not known. SGLT inhibition reduces NHE3 activity and may contribute to increased luminal Na+ concentrations. NaCl detected at the macula densa and via tubuloglomerular feedback (TGF) results in decreased single nephron glomerular filtration rate (SNGFR). Experimental evidence indicates that SGLT2 inhibition may inhibit the diabetes-induced upregulation of renal gluconeogenesis. Increased luminal glucose may facilitate intracellular urate exchange via GLUT9. This promotes uric acid urinary excretion reducing serum levels in diabetes. Diuresis and calorie loss result in reductions in body weight and blood pressure, translating into improved insulin sensitivity and reduced arterial stiffness. Finally, increased urinary excretion of glucose lowers hyperglycaemia and alleviates glucotoxicity at multiple tissue sites including benefits for insulin secretion.
It is anticipated that lowering blood glucose levels with SGLT inhibition will reduce the filtered glucose load and protect both the vascular and basolateral tubular renal components from hyperglycaemia-induced damage. As evidence for this, only when blood glucose levels were substantially lowered with SGLT2 blockade, the kidneys were protected from injury in preclinical models of diabetes; these studies argued against (1) a primary role of SGLT2-mediated glucose uptake in the early diabetes-induced renal growth, inflammation and injury and (2) increased luminal glucose downstream of the early proximal tubule as a potentially adverse consequence for the kidney upon SGLT2 inhibition in diabetes.44,47,78 Micropuncture studies in STZ-induced diabetic rats showed that proximal reabsorption was greater for a given level of single nephron GFR, and the Na+-Cl−-K+ concentration at the macula densa was reduced, suggesting primary proximal tubular hyperreabsorption and the physiology of tubuloglomerular feedback as the main driver for glomerular hyperfiltration in early diabetes.79,80 Application of the non-selective SGLT inhibitor, phlorizin, into the Bowman’s space normalized the Na+-Cl−-K+ concentration at the macula densa and reduced glomerular hyperfiltration without changing blood glucose levels. 79 Lowering glomerular hyperfiltration may likely protect against the development of diabetic kidney disease for the subset of patients who experience glomerular hyperfiltration. Importantly, and consistent with animal studies,44,47,79,81 an 8-week study in type 1 diabetic patients revealed that empagliflozin reduced GFR in hyperfiltering patients (>135 mL/min/1.73 m2). 61 For further discussion on the effects of SGLT2 inhibition on GFR, see Vallon. 82 Ongoing and future research will determine the specific renal adaptations to SGLT2 inhibitors and provide further insight into their long-term safety.
Hypertension is a common co-morbidity in individuals with diabetes, and any therapy that can lower blood pressure would be an effective strategy to prevent kidney disease and cardiovascular mortality. 83 SGLT2 inhibition causes a reduction in systolic blood pressure by ~3–6 mmHg in type 2 diabetes, which is not completely accounted for by the associated reduction in body weight. 84 A small to modest natriuretic and osmotic diuretic effect of SGLT2 inhibition is expected given the contribution of SGLT2 to the retention of sodium, glucose and thereby water. Additionally, a positive interaction between the SGLTs and Na+/H+ exchanger-3 (NHE3), inhibition of NHE3 with phlorizin and the phosphorylation of NHE3 by empagliflozin, at sites associated with reduced NHE3 activity, have been described.85–87 Given that the NHE3 in the early proximal tubule is responsible for up to 30% of fractional sodium reabsorption, its potential downregulated activity upon SGLT2 inhibition may contribute to the natriuresis and subsequent GFR and blood pressure lowering, a hypothesis that requires further studies. The natriuresis does not persist because a new steady state is established, whereby reduced blood pressure and increased activity of the renin angiotensin system stimulate renal Na+ retention via alternate pathways and compensate for the natriuretic effect of the drug.
Benefits in the context of lowering cardiovascular risk factors such as body weight, insulin resistance, renal hyperfiltration, hypertension, arterial stiffness and daily insulin dose (in part due to improvements in pancreatic insulin secretion 88 ) have been reported with SGLT2 inhibition in patients with type 1 and/or type 2 diabetes.56,58,60,62,67 Reductions in systolic blood pressure and body weight may occur as a result of, at least initially, osmotic diuresis and natriuresis upon SGLT2 inhibition. 73 Notably, two-thirds of the weight loss observed in type 2 diabetic patients in response to dapagliflozin treatment for 24 weeks was attributed to reductions in fat mass (visceral and subcutaneous), reflecting significant metabolic adaptations to sustained glucosuria. 89 In diabetic mouse studies, SGLT2 deletion improved glucose-stimulated insulin secretion which was associated with preserved islet mass; probably the result of reduced glucose toxicity. 88 Further research is necessary to better define the mechanisms underlying the wide-ranging pleiotropic benefits associated with targeting SGLT2. With respect to cardiovascular outcomes, a meta-analysis of various Phase II and III clinical trials ranging from 12 to 52 weeks in duration compared dapagliflozin with traditional agents and reported a hazard ratio of 0.67 [95% confidence interval (CI): 0.42–1.08] for a composite cardiovascular end-point including vascular death, non-fatal myocardial infarction or stroke and unstable angina. 90 Cardiovascular data from long-term trials are expected to emerge within the next few years (ClinicalTrials.gov Identifier: NCT01131676, NCT01730534, NCT01032629; see also article by Inzucchi et al., this issue).
Conclusion
Effective glycaemic control in diabetes is important to minimize the progression of the disease and the risk of potentially devastating complications. Diabetes increases the renal expression of SGLT2 and thereby may contribute to increased glucose reabsorption. Inhibition of SGLT2 lowers renal glucose reabsorption and blood glucose levels via a predominantly insulin-independent mechanism. Therefore, provided renal filtration remains adequate, these drugs may be effective at all stages of type 2 diabetes and perhaps also in type 1 diabetes. SGLT2 inhibition unmasks the transport capacity of SGLT1, which explains why renal glucose reabsorption is maintained at ~50% when SGLT2 is inhibited under normal glucose conditions. Together with the role of SGLT1 in intestinal glucose reabsorption, this provides a rationale for the use of dual SGLT1/SGLT2 inhibitors to maximize the degree of blood glucose lowering. Certainly, the potential for intestinal malabsorption and effects in other SGLT1-expressing organs need to be carefully addressed, as do the downstream effects on kidney function. SGLT2 inhibitors induce pleiotropic actions, including reduced body weight, glomerular hyperfiltration and hypertension that may translate into improved cardiovascular outcomes in diabetes. Long-term clinical studies are ongoing and will determine whether SGLT2 inhibitors have a safety profile and exert cardiovascular benefits that are superior to more traditional agents and help to further define their role in anti-diabetic therapy.
Footnotes
Acknowledgements
L.A.G., E.M.W. and V.V. provided the concept for the article, contributed to interpretation of data, drafting and critically revising for important intellectual content and provided final approval of the version to be submitted for publication.
Declaration of conflicting interests
V.V. was supported by investigator-initiated research grants from Bristol-Myers Squibb, AstraZeneca and Boehringer Ingelheim, Biberach. V.V. serves as a consultant for Boehringer Ingelheim and Janssen Pharmaceutical. E.M.W. serves on a Boehringer Ingelheim Advisory Board and has been supported by investigator-initiated research grants from Boehringer Ingelheim and Janssen Pharmaceuticals.
Funding
L.A.G. was supported by a Heart Foundation Postdoctoral Fellowship (Australia). E.M.W. was supported by the National Institutes of Health (R01DK19567). V.V. was supported by the National Institutes of Health (R01DK56248, R01HL094728 and P30DK079337), the American Heart Association (GRNT3440038) and the Department of Veterans Affairs.