
Abstract
Objective:
C-reactive protein (CRP) levels in diabetes predict cardiovascular events. Also, human CRP (hCRP) exacerbated the proinflammatory, pro-oxidant and procoagulant states in a spontaneous model of type 1 diabetes mellitus (T1DM), the biobreeding (BB) rat. Since there is a paucity of data examining the role of CRP on endothelial dysfunction in animal models of diabetes, we tested this hypothesis in the diabetic BB rat.
Methods:
Diabetic BB rats (n = 4 per group) were injected with human serum albumin (HSA) or hCRP [hCRP = 20 mg/kg body weight; intraperitoneal (IP)] for three consecutive days. The rats were euthanized on day 4. Biomarkers that were assayed included endothelin-1 (ET-1), soluble intracellular adhesion molecule-1 (sICAM-1), Von Willebrand factor (vWF) and 6-keto prostaglandin F1-alpha (6-keto PGF1-α) in plasma.
Results:
hCRP administration resulted in a significant increase in plasma levels. Furthermore, hCRP-treated rats had significantly increased circulating levels of ET-1 (1.12 ± 0.6 pg/mL versus 0.4 ± 0.21 pg/mL), vWF (45 ± 2.4 ng/mL versus 34 ± 7 ng/mL) and sICAM-1 (41 ± 3 ng/mL versus 34 ± 3.4 ng/mL) compared to HSA-treated rats (p < 0.05). There was no significant effect on 6-keto PGF1-α levels.
Conclusion:
Hence, in this preliminary report, we make the novel observation that hCRP induces endothelial dysfunction in a spontaneous model of T1DM, and this could have implications for the vascular complications in diabetics.
Introduction
C-reactive protein (CRP), the prototypic marker of inflammation, predicts cardiovascular disease events and cardiovascular mortality in diabetes.1–3 Type 1 diabetes mellitus (T1DM) is a proinflammatory state typified by increased CRP levels, endothelial dysfunction and increasing risk of cardiovascular disease.4,5 The biobreeding (BB) rat spontaneously develops T1DM via a T-cell-mediated autoimmune destruction in pancreatic beta cells and is a valid model of T1DM to test the effects of human CRP (hCRP).6,7
Previously, we reported that hCRP administration compared to human serum albumin (HSA) exacerbates the proinflammatory, pro-oxidant and procoagulant states of diabetes, largely by activating macrophages in the diabetic BB rat.7,8 In these previous reports, we demonstrated increase in plasma plasminogen activator inhibitor-1 (PAI-1) levels. Since PAI-1 is derived from many sources including endothelium and we showed that CRP induces PAI-1 secretion from endothelial cells,9,10 we speculated that this could also be evidence of endothelial dysfunction. Furthermore, many groups have reported that CRP induces endothelial dysfunction both in vitro and in vivo in non-diabetic models.11,12 Hence, we hypothesized that CRP could possibly exacerbate endothelial dysfunction under diabetic conditions. In this study, we report on the effects of hCRP on additional biomarkers of endothelial dysfunction in diabetic BB rats.
Materials and methods
The protocol was approved by the Animal Committee of University of California at Davis. Male biobreeding diabetes-prone (BBDP) rats (125–150 g; BRM Inc., Boston, MA, USA) were housed in a controlled sterile environment (pathogen-free conditions) under a 12-h light/12-h dark cycle, and diabetes was confirmed by measuring tail vein blood glucose levels using a commercial glucometer (ACCU-CHEK; Roche Diagnostics, Indianapolis, IN, USA) as described previously.7,8 The rats were considered diabetic when blood glucose levels exceeded 300 mg/dL on three consecutive days. Animals were maintained in hyperglycaemic condition (300–450 mg/dL) for 2 weeks by inserting subcutaneous insulin pellets (Linplant; LinShin, Toronto, ON, Canada) (1–2 U/day) to prevent decompensation and weight loss.
In this study, CRP was purified from human pleural or ascites fluid as reported previously.7,8,13 Endotoxin was removed from the purified CRP preparation using detoxigel columns (Pierce Biochemicals, Rockford, IL, USA) and had <0.125 EU/mL (<12.5 pg/mL) of endotoxin measured by the Limulus assay (BioWhittaker, East Rutherford, NJ, USA). CRP protein was further concentrated (Amicon, Inc., Beverly, MA, USA), dialyzed, sterile filtered and stored at 4°C. This procedure resulted in a CRP preparation that induced proinflammatory effects in TLR4 knockdown endothelial cells and macrophages using small interfering RNA (siRNA) technology.14,15 TLR4 is the receptor via which endotoxin mediates its effect. All the reagents and buffers used in this study were tested and had endotoxin <0.125 EU/mL. Sterile-filtered, fatty acid–free and endotoxin-free HSA prepared in Tris-HCL buffer with 2 mM calcium was used as the control protein, as reported previously,7,8,13 to compare it with inflammatory effects of CRP on endothelial dysfunction.
After 2 weeks of persistent hyperglycaemia, hCRP or HSA was administered by intraperitoneal (IP) injection (20 mg/kg body weight for 3 days), as described previously, to BBDP rats.7,8,13 The rats were euthanized on day 4, and plasma was collected for measurement of biomarkers of endothelial dysfunction. Plasma levels of hCRP were measured using enzyme-linked immunosorbent assay (ELISA), which recognizes only hCRP and not rat CRP as described previously.7,8,13
Endothelial dysfunction biomarkers
Plasma levels of endothelin-1 (ET-1) and soluble intracellular adhesion molecule (sICAM-1) were quantitated by specific sandwich enzyme immunoassays from R&D Systems (Minneapolis, MN, USA). The intra-assay coefficient of variation (CV) for ET-1 and sICAM-1 ELISA was 4.4% and 7%, respectively.
Von Willebrand factor (vWF) in rat plasma was measured using a sandwich enzyme immunoassay kit from USCN (Houston, TX, USA). The intra-assay CV for vWF ELISA was 4.9%.
As a read-out of prostacyclin synthase activity, we quantitated 6-keto prostaglandin F1-alpha (6-keto PGF1-α) concentrations in plasma samples by competitive enzyme immunoassay (EIA) using reagents from Cayman Chemicals (Ann Arbor, MI, USA), where the amount of 6-keto PGF1-α tracer binding to rabbit antiserum was inversely proportional to concentration of 6-keto PGF1-α in the rat plasma. The intra-assay CV for 6-keto PGF1-α ELISA was 3.6%.
All assays were performed in duplicate. The comparisons between group means were analysed using GraphPad Prism. The experimental results are presented as the mean ± standard deviation (SD). t-tests were used to compute differences in the variables, and the level of significance was set at p < 0.05.
Results
The average glucose levels of hCRP-treated (n = 4) and HSA-treated (n = 4) diabetic BB rats were 324 ± 33 and 359 ± 50 mg/dL, respectively. Administration of hCRP to diabetic rats significantly increased circulating levels of hCRP (26 ± 6 mg/L) 24 h after the last injection, confirming that hCRP entered the systemic circulation unlike in HSA-injected BB rats in which hCRP levels were undetectable. There was a significant increase in levels of vWF in CRP-injected rats compared to HSA-treated rats (p < 0.05) (Figure 1(a)). ET-1 was also found to be significantly elevated in hCRP-administered rats versus HSA-treated rats (p < 0.05) (Figure 1(b)). Similarly, sICAM-1 levels were also significantly increased in CRP-injected rats compared to HSA controls (p < 0.05) (Figure 1(c)). Circulating 6-keto PGF1-α levels were not significantly decreased in plasma with hCRP administration [721 (404–1055) pg/mL] compared to HSA-treated BB rats [2214 (1190–6340) pg/mL (p = 0.2)].
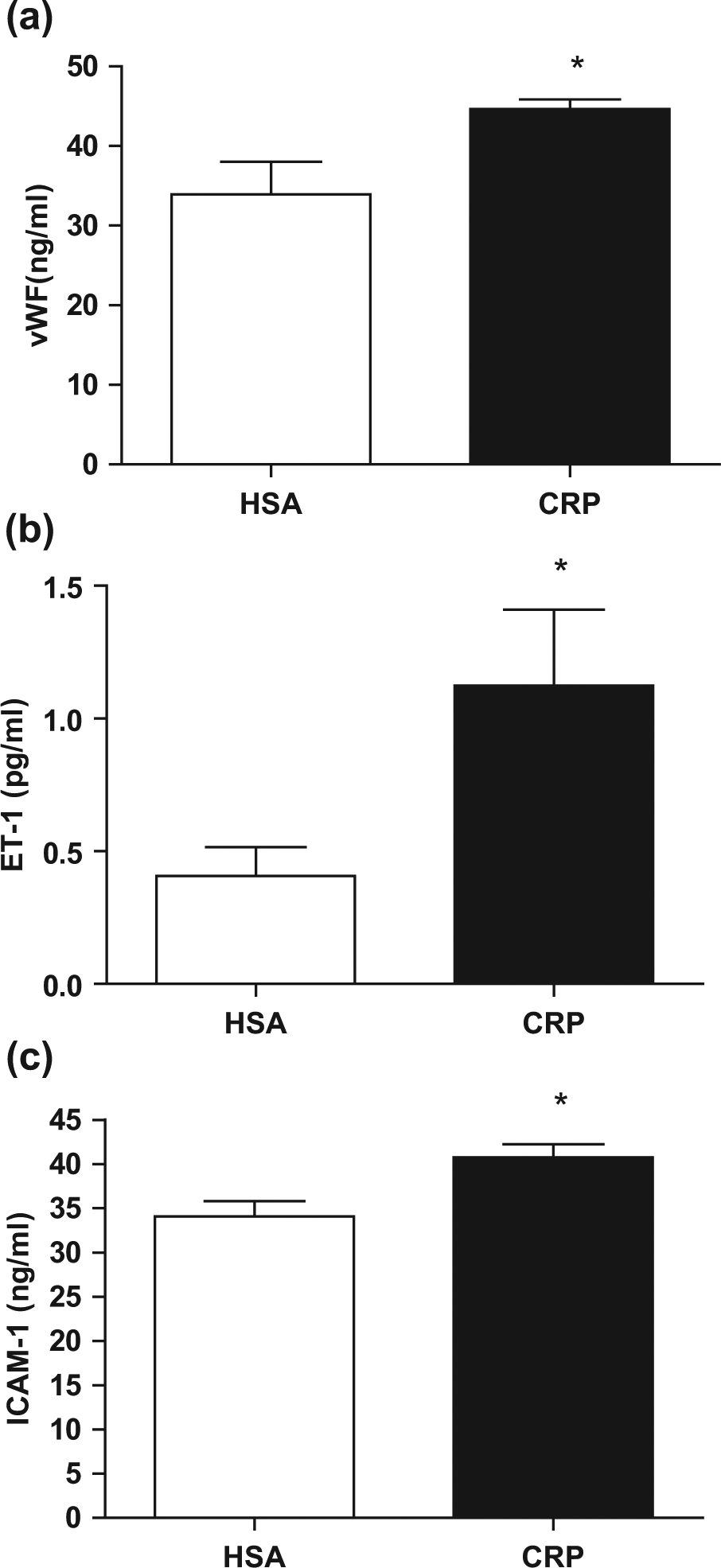
(a) Plasma vWF in hCRP-treated (n = 4) and HSA-treated (n = 4) diabetic BB rats (*represents p < 0.05 compared to HSA-treated diabetic BB rats), (b) plasma ET-1 levels in hCRP-treated (n = 4) versus HSA-treated (n = 4) diabetic BB rats (*represents p < 0.05 compared to HSA-treated diabetic BB rats) and (c) plasma sICAM-1 levels in hCRP-treated (n = 4) versus HSA-treated diabetic BB rats (n = 4) (*represents p < 0.05 compared to HSA-treated diabetic BB rats).
Discussion
Increased CRP is a known risk marker for predicting cardiovascular disease.1–3 Previous studies have suggested that CRP promotes increased monocyte/macrophage activity, inflammation and endothelial dysfunction.11,12 Furthermore, a relationship between vascular dysfunction and CRP levels has been reported. 11 We hypothesized that CRP exacerbates vascular dysfunction in T1DM using the diabetic BB rat as our model.
In our previous report on the BB rat, we convincingly detailed that hCRP accentuates macrophage activity as evidenced by increased expression of protein kinase C (PKC)-alpha, PKC-delta, p47phox and increased NFκB activity and biomediators of inflammation compared to HSA control. 7 Hence, on stored frozen plasma from the previous study, we decided to investigate other biomarkers of endothelial dysfunction, given the known effects of CRP on endothelial dysfunction in non-diabetic models 16 and our finding of increased PAI-1 levels. Various reports point towards a role of CRP in mediating endothelial dysfunction including increase in ET-1, PAI-1, cell adhesion molecules (CAM) [such as ICAM-1, vascular cell adhesion molecule-1 (VCAM-1) and E-selectin], macrophage colony stimulating factor (M-CSF) and monocyte chemotactic protein-1 (MCP-1) and decrease in levels of prostacyclin, tissue plasminogen activator (tPA) and endothelial nitric oxide synthase (eNOS) activity.11,17
In this report, we found significantly increased levels of vWF, ET-1 and sICAM-1 in hCRP-treated diabetic BB rats compared to HSA controls. It needs to be pointed out that our carefully purified CRP exerts proinflammatory effects on TLR4 knockdown cells suggesting that the effects we demonstrate are not due to endotoxin contamination. In this study, we report for the first time, significantly increased vWF levels in hCRP-treated diabetic BB rats compared to controls. Wynants et al. 18 previously demonstrated that CRP significantly increased secretion of vWF, ET-1 and adhesion capacity in endothelial cells from patients with chronic thromboembolic pulmonary hypertension (CTEPH) further underscoring the role of CRP in promoting endothelial dysfunction.
Verma et al. 19 also reported previously that CRP significantly induces release of ET-1 and adhesion molecules (ICAM-1, VCAM-1) in human venous endothelial cells supporting CRP-mediated endothelial dysfunction. Guan et al. 20 found significantly increased expression of ET-1, angiotensin receptors (AT-1) and endothelin type A receptors as well as decreased expression of eNOS and AT-2 receptors in male Wistar rats administered adenoassociated virus (AAV)-hCRP compared to controls, suggesting a role of hCRP in promoting impaired endothelial function. The CRP-induced increased sICAM-1 levels in diabetic BB rats also accord with the published literature.11,12
Endothelial vasodilatation is mediated by both prostacyclin and nitric oxide.11,17 It has been demonstrated previously that hCRP decreases eNOS activity and significantly decreases release of prostacyclin in vitro in human aortic endothelial cells (HAEC) contributing to endothelial dysfunction. 21 Hein et al. 22 showed significantly decreased eNOS activity in vivo in Sprague Dawley rats injected with hCRP. However, we failed to show significant decrease in prostacyclin release in CRP-treated diabetic BB rats versus controls in this study. This could be due to the small sample size. A definite limitation of this brief report is the small sample size.
Previously, it has been shown that CRP induces endothelial dysfunction in the non-diabetic milieu by uncoupling eNOS, nitrating prostacyclin synthase and activating NFκB.21–23 In future studies, we will also investigate these and other mechanisms in this model of T1DM. Thus, we clearly confirm the data from many groups that hCRP induces endothelial dysfunction especially in a diabetic model as evidenced by significant alterations in three of our four biomarkers. Thus, CRP could be a potential target for therapy to reduce diabetic vascular complications.
Footnotes
Acknowledgements
I.J. and S.D. designed experiments, analysed data and helped with manuscript preparation. H.K. helped with performing assays, data analysis and manuscript preparation. G.S. helped with performing assays and manuscript submission.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
This study was supported by National Institutes of Health (grant no. ROI-HL074360).