
Abstract
In healthy individuals, the vascular endothelium regulates an intricate balance of factors that maintain vascular homeostasis and normal arterial function. Functional disruption of the endothelium is known to be an early event that underlies the development of subsequent cardiovascular disease (CVD) including atherosclerosis and coronary heart disease. In addition, the rising global epidemic of type 2 diabetes is a significant problem conferring a significantly higher risk of CVD to individuals in whom endothelial dysfunction is also notable. This review first summarises the role of endothelium in health and explores and evaluates the impact of diabetes on endothelial function. The characteristic features of insulin resistance and other metabolic disturbances that may underlie long-term changes in vascular endothelial function (metabolic memory) are described along with proposed cellular, molecular and epigenetic mechanisms. Through understanding the underlying mechanisms, novel targets for future therapies to restore endothelial homeostasis and ‘drive’ a reparative cellular phenotype are explored.
Introduction
Largely owing to the vast quantities of energy-dense food consumed in the Western world, coupled with increasingly sedentary lifestyles, diabetes mellitus (DM) is considered to be a public health problem of epidemic proportions.1,2 In the United Kingdom alone, 2.9 million people are diagnosed with the disease, with an estimated 850,000 more undiagnosed. 3 DM is recognised as an independent risk factor for cardiovascular disease (CVD), 4 even when under glycaemic control. With up to 75% of mortality in diabetic patients arising from CVD, 5 this is an issue of utmost importance. Endothelial cell (EC) dysfunction is associated with both diabetes and the pathogenesis of CVD. 6 Establishing the causative mechanisms linking these factors could provide essential insights and inform novel therapeutics.
The aim of this review is to explore the relationship between DM and EC function/dysfunction, focussing predominantly on cellular and molecular mechanisms as opposed to clinical manifestations.
Physiological role of endothelium
Forming a vast interface between the blood and surrounding tissues, the endothelium forms a monolayer comprising the innermost lining of blood vessels. Advances over recent decades have revealed the complexity of this semi-permeable membrane and its key role in maintaining vascular homeostasis. 7 While facilitating the passage of substances such as nutrients and leukocytes across the vessel wall, the endothelium secretes numerous mediators necessary for normal vascular functioning including those that regulate vascular tone, coagulation, modulate immune responses and control vascular cell growth. 8
Arguably, the most significant endothelium-derived mediator is nitric oxide (NO), which plays multiple roles in preventing atheroma formation. 9 Produced by the action of endothelial nitric oxide synthase (eNOS), NO diffuses into neighbouring vascular smooth muscle cells (SMC), activating guanylyl cyclase and producing cyclic guanosine monophosphate (cGMP) and activating kinases responsible for vascular relaxation. 8 Vasodilation via NO synthesis is well documented in vitro and in vivo.10–12 Additionally, NO inhibits platelet aggregation, SMC proliferation and nuclear transcription of leukocyte-adhesion molecules including vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM). Arterial wall shear stress 13 and coupling of agonists such as acetylcholine and bradykinin to endothelial-bound receptors 8 are potent stimuli for NO synthesis. Although these stimuli act via different intracellular mechanisms, each increases NO production via enhanced expression of eNOS. Other important vasodilatory factors are endothelium-derived hyperpolarizing factor (EDHF) and prostacyclin (PGI2), the latter also playing a role in platelet inhibition. 8
Conversely, the endothelium is able to secrete the potent vasoconstrictor endothelin (ET-1), which through its pro-inflammatory and mitogenic effects augments the pathogenesis of CVD. 14 Other endothelial-derived vasoconstrictors include prostaglandin H2 (PGH2), thromboxane A2 (TXA2) and reactive oxygen species (ROS). EC-bound angiotensin-converting enzyme (ACE) catalyses production of the vasoconstrictive angiotensin II, and ACE inhibitors are commonly used to decrease blood pressure. 15 Under physiological conditions, the endothelium maintains a fine balance between anti- and pro-thrombotic states. While NO, prostacyclin and thrombomodulin favour blood fluidity and are dominant under basal conditions, ‘activated’ endothelium drives an opposing pro-thrombotic state. Under these conditions, plasminogen activator inhibitor-1 and thromboxane, together with von Willebrand Factor (vWF), a key constituent of the coagulation cascade, are deleterious.8,16
Through NO production, the endothelium plays a crucial role in preventing leukocyte adhesion; however, when ‘activated’, expression of adhesion molecules aid the passage of leukocytes across the vascular wall promoting a pro-inflammatory environment. Under these conditions, EC produce pro-inflammatory cytokines, including tumour necrosis factor-alpha (TNF-α), which further augment inflammation. 17
Finally, the endothelium plays a key role in angiogenesis initiated by tissue growth factors, particularly vascular endothelial growth factor (VEGF). The VEGF receptor is coupled to activation of mitogen-activated protein kinase (MAPK) signalling, promoting angiogenesis by increasing nuclear transcription of relevant genes. This mechanism is counteracted by the anti-angiogenic factors angiostatin and thrombospondin, which suppress angiogenesis unless required. 8
Endothelial dysfunction
Endothelial dysfunction refers to inability of the endothelium to regulate vascular homeostasis, and essentially describes ‘tipping’ of the physiological balance in favour of vasoconstrictive, pro-inflammatory and pro-thrombotic effects 5 that promote atherosclerosis. As such, abnormalities in endothelial function are detected early in the development of CVD, often before symptoms are clinically evident. 16
Given the vast range of vasoprotective effects of NO, the term endothelial dysfunction generally refers to reduced NO bioavailability, through decreased eNOS expression. 16 Procedures employed to evaluate endothelial dysfunction measure blunted vasodilatory responses to certain agonists. 5 Indeed, by this method, impaired endothelial-dependent vasodilation in patients with confirmed CVD,18–21 and importantly in those who carry risk factors for future CVD events, 22 has been demonstrated. This suggests that endothelial dysfunction is the commonality by which these risk factors, particularly those associated with the metabolic syndrome, lead to CVD development. This point is underscored by reports that endothelial dysfunction appears to reverse when lifestyle changes and modulatory drug therapies are implemented. 23 DM is one of the aforementioned risk factors whose association with endothelial dysfunction will now be evaluated.
Endothelial dysfunction in DM
Current evidence links endothelial dysfunction to type 1 DM (T1DM) and type 2 DM (T2DM), through demonstration of impaired endothelial-dependent vasodilation.24–26 Despite many proposed mechanisms for this relationship, the definitive pathogenesis remains unclear, possibly because diabetes patients usually display multiple homeostatic imbalances alongside the typically described hyperglycaemia. Such disturbances induce endothelial dysfunction independently of the presence of diabetes, indicating a multifactorial aetiology rather than hyperglycaemia per se.27–29 The following sections will evaluate these factors.
Selective insulin resistance
In addition to its role in glucose homeostasis, insulin activates intracellular signalling pathways critical for maintenance of healthy endothelium, the most vasoprotective of which is the phosphoinositide-3 kinase (PI3-K)/Akt pathway (Figure 1). Through this pathway, enhanced eNOS expression and activation (phosphorylation) are mediated.30,31 In contrast, activation of the MAPK/extracellular signal-regulated kinase (ERK) pathway, promotes expression of ET-1 and a less beneficial outcome of cellular proliferation. 32 In physiological circumstances, the PI3-K pathway predominates to regulate normal vasomotor control, 33 but it is well accepted that in insulin resistance that characterises T2DM, there is a selective deficiency of the PI3-K pathway, while signalling via the MAPK pathway is unaffected.34,35 Moreover, this selective resistance promotes hyperinsulinaemia, resulting in further stimulation of MAPK signalling. 36 The outcome therefore is that of pro-atherogenic signalling that is clearly detrimental to the vasculature. 37 This signalling deficiency has been demonstrated experimentally in obese Zucker rats, in which defective PI3-K/Akt signalling consequently resulted in decreased NO bioavailability. 34
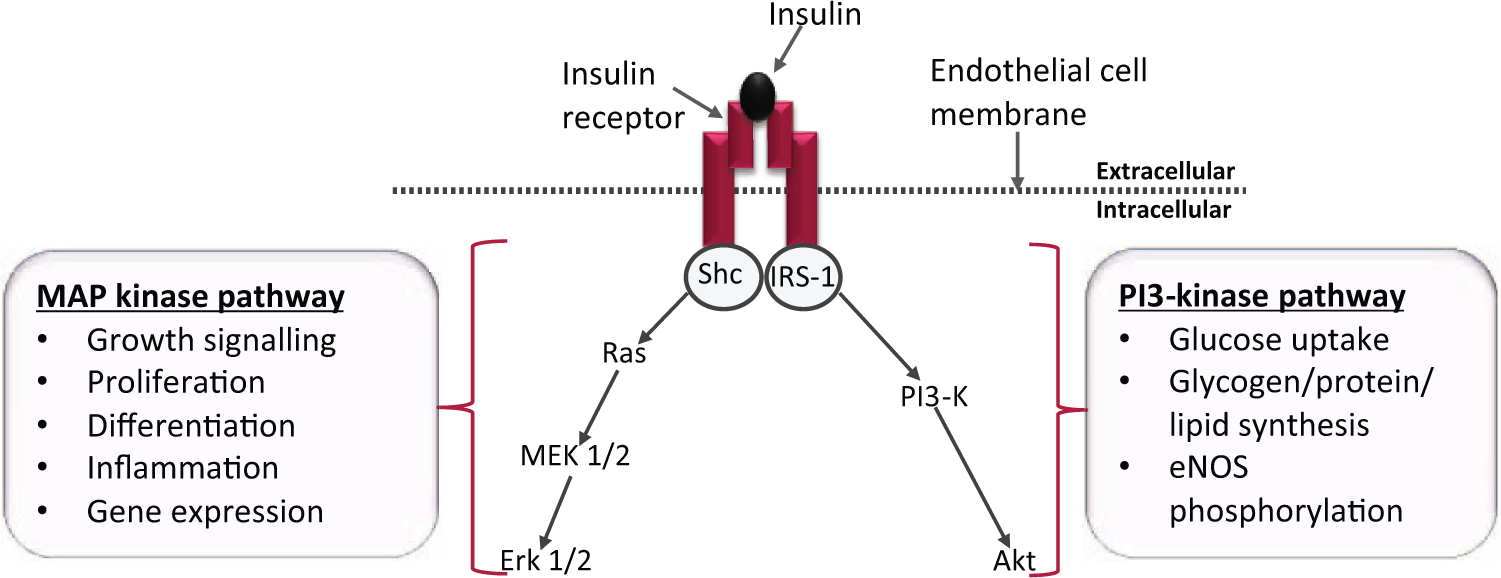
Intracellular insulin signalling pathways: Upon binding to its receptor, insulin initiates two key intracellular signalling pathways. Activation of PI3-Kinase dominates under physiological conditions resulting in a net anti-atherogenic effect. In insulin resistance, selective deficiency in the PI3-kinase pathway leads to a dominant MAP kinase pathway, whose effects are predominantly pro-atherogenic.
Pro-inflammatory signalling
T2DM is associated with a state of chronic systemic inflammation; elevated levels of circulating inflammatory markers are observed in patients with diabetes and obesity38,39 and are believed to confer a pathological EC phenotype. A diverse range of harmful stimuli have been shown to contribute to vascular complications in diabetes. These include pro-inflammatory cytokines, chemokines, adhesion molecules and transcription factors (summarised in Table 1) and have been reviewed recently. 40
Inflammatory components of diabetes complications.

Key categories of inflammatory mediators associated with vascular complications of diabetes, indicating the principal components of each category. There is widespread evidence for inflammatory cytokines, adhesion molecules, chemokines and transcription factors. More recent data support roles for profibrotic cytokines and toll-like receptors that appear to mediate responses to a number of ‘diabetogenic’ stimuli (reviewed in Forbes and Cooper 40 ).
Animal models of obesity have identified adipose tissue as a site of inflammatory cytokine production, with accompanying elevated plasma levels of TNF-α.41,42 TNF-α activates nuclear factor κB (NFκB), a transcription factor able to further stimulate expression of inflammatory genes. 43 NFκB is also activated by free fatty acids (FFA) and the receptor for advanced glycation end-products (RAGE), both of which are characteristic of the diabetic milieu. 16 Evidence suggests that overexpression of either TNF-α or IκB kinase mediates the development of insulin resistance. Obese mice with null mutations in the TNF-α gene display significantly improved insulin sensitivity. 44 Indeed, NFκB that is permanently present in the cell could be viewed as the ‘first responder’ to many inflammatory signals due to rapid activation of its downstream pathways. More recently, new pharmacological roles for the salicylate anti-inflammatory drugs have emerged regarding their ability to decrease insulin resistance via inhibition of IκB kinase. Preliminary results from the TINSAL-T2D (Targeting INflammation Using SALsalate in T2DM) clinical trial advocates their use in glucose-lowering, presumably as a consequence of improved insulin sensitivity. 45 The association of insulin resistance with inflammation implies that the latter adversely impairs the NO-producing PI3-K/Akt pathway, ultimately reducing NO bioavailability. Exposure of cultured EC to TNF-α leads to impaired eNOS expression, 46 inferring that reduced bioavailability of NO nullifies its otherwise significant anti-inflammatory role, thus augmenting formation of inflammatory atherosclerotic lesions.
Oxidative stress
Oxidative stress is commonly implicated as an important unifying mechanism in conferring endothelial injury (see Figure 2). The term refers to the accumulation of ROS such as superoxide anion (O2·−), when production exceeds the capability of the anti-oxidant system to remove them. 47 A key adverse effect of oxidative stress is diminished NO bioavailability, either through direct degradation by ROS, or alterations in the functional capacity of eNOS. Direct reaction of NO with O2·− yields peroxynitrite (ONOO−), a highly potent oxidant capable of altering the functions of multiple intracellular molecules, including those related directly to the NO pathway. 48 Additionally, ONOO− serves to uncouple eNOS from its critical co-factor tetrahydrobiopterin (BH4), resulting in preferential formation of O2·− in the place of NO and hence further exacerbation of the problem. 49 In the setting of insulin resistance, experimental evidence has implicated endogenous ONOO− as a down-regulator of the PI3-K/Akt pathway.50,51
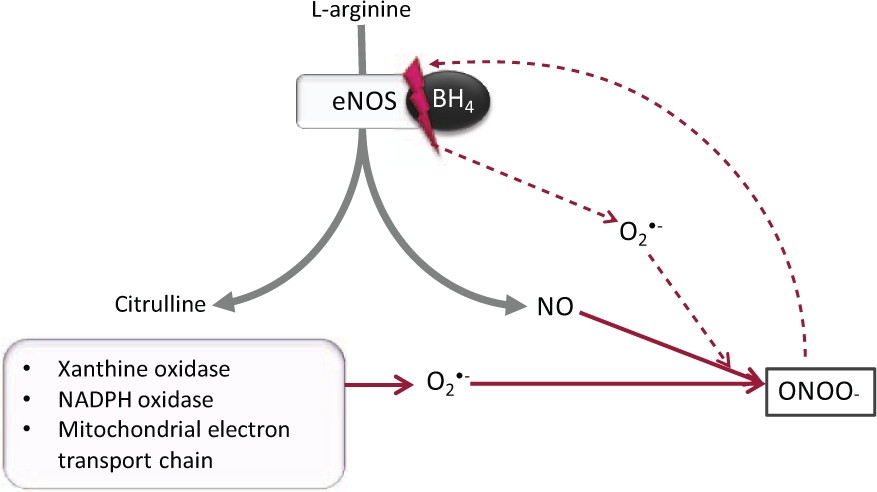
Production of peroxynitrite in endothelial cells: In healthy cells, eNOS catalyses synthesis of NO from
Aside from uncoupled eNOS, significant sources of O2·− include xanthine oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. 47 Mitochondria produce negligible ROS in physiological circumstances, but greatly increase O2·− output when high glucose concentrations increase the proton gradient within the electron transport chain. 52 Recent evidence has also implicated mitochondrial dynamics, namely, increased mitochondrial fission and fragmentation, as contributing factors to ROS production in DM. 53
Anti-oxidant therapy
Anti-oxidant therapies aiming to reduce the detrimental effects of ROS have so far yielded conflicting results. Studies of diabetic vessels in vivo have demonstrated improvements in NO-mediated vasodilation in response to anti-oxidants such as superoxide dismutase (SOD).54,55 Additionally, the anti-oxidant properties of vitamin C have been shown to effectively augment endothelial-dependent vasodilatation in several studies performed on human forearm vessels, in the setting of diabetes and hypertension.56–58 Despite promising experimental data, large-scale clinical trials investigating the effects of anti-oxidant vitamins in patients with diabetes failed to report any benefits.59,60 Although conducting clinical trials is a logical progression following experimental evidence, the discrepancies in these results may indicate that the benefits of anti-oxidant therapies are too short term to affect mortality outcomes. The vastly greater sample size and follow-up period in clinical trials indicates that these results should take precedent when interpreting the effects of anti-oxidant therapy. However, further experimental evidence regarding the longer term outcomes of anti-oxidant therapies is required.
Protein kinase C
Protein kinase C beta (PKCβ) is an endothelial isoform of the serine/threonine kinase family. A host of evidence implicated this enzyme as a chief contributor to endothelial dysfunction observed in DM. 61 PKCβ is activated by diacylglycerol (DAG), which is typically produced subsequent to ligand-receptor binding. 62 However, in conditions of high circulating FFA and hyperglycaemia, a novel route for the chronic activation of DAG has been proposed, via de novo synthesis from glucose. 63 Significantly increased levels of PKC and DAG have been identified in animal models of diabetes. 63 Upon activation, PKC induces several intracellular effects (Figure 3), many of which have been experimentally identified using PKC inhibitors such as ruboxistaurin.43,64 Such alterations include amplified expression of ET-1, VCAM and ICAM. 62 PKC also activates vascular NADPH oxidase, a significant source of O2·− and subsequent oxidative stress in the endothelium. Indeed, inhibition of PKC has been shown to diminish O2·− in the diabetic vasculature. 65 A trial in healthy humans indicated that PKC inhibition prevented hyperglycaemia-induced impairment of endothelial-dependent vasodilation. 66 Disappointingly, a later trial by the same authors showed that this benefit was not apparent in T2DM patients, 67 suggesting that further work is required to delineate the role of PKC in T2DM. Additional effects of PKC in the endothelium include activation of NFκB, alterations in eNOS expression and inhibition of the PI3-K signalling pathway. 43
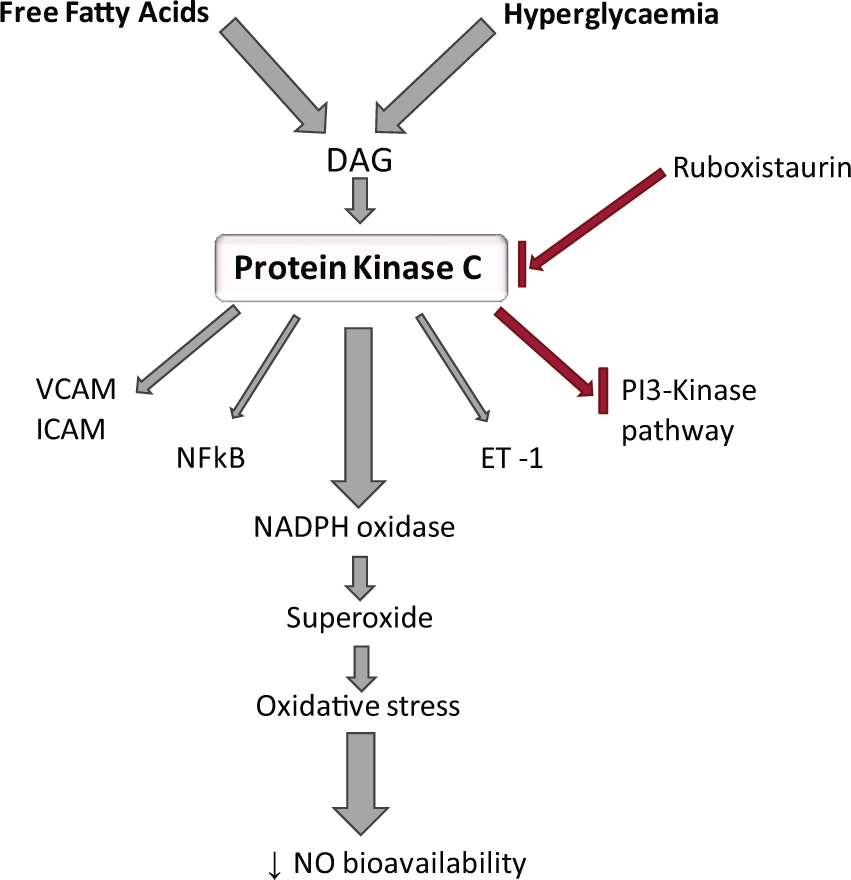
Activation and effects of protein kinase C (PKC): High levels of free fatty acids and hyperglycaemia activate DAG, which in turn activates PKC. This increases expression of ET-1, VCAM, ICAM, NFκB and NADPH oxidase. NADPH oxidase activation results in decreased NO bioavailability. PKC may have a role in PI3-kinase pathway inhibition. Ruboxistaurin inhibits PKC activation.
Hyperglycaemia
The hallmark of DM, chronic hyperglycaemia, has been implicated in the development of endothelial dysfunction via four principal mechanisms: PKC activation, activation of the hexosamine and polyol pathways and formation of advanced glycation end-products (AGEs). 68 These pathways are believed to mediate vascular dysfunction through the unifying mechanism of ROS overproduction, most notably increases in O2·−. 69 Briefly, the polyol and hexosamine pathways display low affinity for glucose in the physiological state; however, intracellular hyperglycaemia leads to shunting of glucose through these pathways, with resultant increases in PKC activation, changes in gene expression and protein function. 68 AGEs initially arise from intracellular hyperglycaemia and subsequent non-enzymatic reactions, causing alterations in both intra- and extracellular proteins, as well as modifications to the extracellular matrix. 70 Additionally, activation of RAGE on EC influences ROS production and leads to NFκB activation, which induces atherogenic gene expression.71,72 Collectively, these mechanisms have been shown to increase the proton gradient across the inner mitochondrial membrane, consequently increasing O2·− as previously discussed. SOD-enabled inhibition of mitochondrial ROS is capable of inhibiting increases in all these mechanisms. 68
Metabolic memory: role of epigenetics
The mechanisms described suggest routes by which environmental factors such as diet-induced hyperglycaemia may induce endothelial dysfunction, and it would be easy to believe that correcting these imbalances would promptly normalise endothelial function. This is not the case. Indeed, follow-up trials succeeding the Diabetes Control and Complications Trial (DCCT) and the United Kingdom Prospective Diabetes Study (UKPDS) propose the existence of a so-called metabolic memory, in which the effects of either prolonged or transient changes in glycaemia persist long after these have been re-adjusted.73–75 Not only does this suggest that intensive glycaemic control cannot completely reverse pre-established hyperglycaemia-induced vascular complications, but importantly showed that the benefits of intensive therapy were maintained for many years despite a return to often inferior glycaemic control. 73 Such observations have been confirmed in vitro and in vivo, including EC-specific studies.76–78 Together, these data imply that heritable alterations in cell phenotype may be induced by diabetogenic stimuli, particularly hyperglycaemia. While this was once thought to be due to mutations in mitochondrial DNA, 68 more recent evidence has implicated epigenetic mechanisms as the underlying cause of this phenomenon–alterations in gene expression without changes in DNA sequence. 79 Indeed, epigenetic studies are shedding new light on how environmental factors influence gene expression and the associated susceptibility to T2DM and increased CVD. 79 Three highly connected pathways provide the foundation for epigenetic theories, namely, post-translational histone modifications, DNA methylation and non-coding RNA-based mechanisms, detailed studies of which are clearly warranted to investigate endothelial-specific mechanisms that may inform novel therapeutic targets.
Histone modifications
Post-translational modifications of histones in chromatin alter the conformation of DNA, providing or denying access to specific sites that enhance or retard gene transcription, respectively. Histone modifications (e.g. acetylation/deacetylation; methylation/demethylation) are regulated by a family of enzymes that are responsible for their transfer [histone acetyltransferases (HATs), histone methyltransferases (HMTases)] or removal [histone deacetylases (HDAC), histone demethylases (HDM)]. 79 Several studies have been performed specifically in ECs, where changes in the expression and activity of these enzymes have been linked to diabetic complications. For example, specific histone methylations are associated with the chronic inflammatory phenotype in EC exposed to high glucose concentrations. 80 Transient exposure of human and bovine endothelium to hyperglycaemia led to transcriptional activation of inflammatory NFκB-dependent promoters that were sustained even following restoration of normoglycaemia.80,81
DNA methylation
Studies examining the role of DNA methylation in diabetes and vascular complications are few, although altered patterns of DNA methylation have been associated with atherosclerosis 82 and in the endothelium itself. 83 Both global and gene-specific methylation is believed to contribute to the metabolic memory of diabetic complications. Covalent methylation of cytosine residues is a stable DNA modification that persists throughout experimental processing, 84 providing a robust platform on which to base future investigations. Akin to modifications in histones, DNA methylation can confer persistent alterations in gene expression and a clear understanding of the complex mechanisms involved will be of immense value. High fat feeding of obese rats has been reported to induce hypermethylation of particular genes, 85 so it is reasonable to consider that this phenomenon may also exist in human diabetic individuals, and specifically at the level of the endothelium.
microRNAs
Another area of intense interest in which gene expression can be post-transcriptionally modified is by microRNAs (miRs), the most widely studied of the non-coding RNAs. miRs are short RNAs that inhibit expression of a range of target proteins through mRNA degradation or translational repression. As regards ECs, in vitro studies have exposed a complex network of miRs with regulatory roles in proliferation and migration (miR-320, 86 miR-503, 87 miR221/22288–90), inflammation (miR-10a, 91 miR-126 92 ) and angiogenesis (miR221/222, 93 miR-126, 94 miR-210, 95 miR-21 96 ). These roles may be divergent in physiological circumstances, for example, specific miRs are capable of exerting protective, pro-angiogenic effects,94,95 while others confer more unfavourable anti-angiogenic properties.93,96 Furthermore, a distinct signature of miR dysregulation has been proposed to exist in DM, 97 whereby high glucose conditions may induce over or under expression of detrimental or beneficial miRs, respectively, thereby contributing to the development of vascular complications. In such a way, upregulation of miR-320 was observed in EC exposed to hyperglycaemia, with a resultant reduction in cellular proliferation and migration. 86 Importantly, inhibition of the overexpressed miRs restored EC regenerative functions. 86
The stable and consistent levels of plasma miRs in healthy individuals has identified them as potential biomarkers for diseases such as T2DM, 98 not only for evaluating disease progression but also to detect risk of future disease development. In this way, plasma profiling of T2DM patients has revealed altered levels of miRs, which may contribute to a pathophysiological state. 99 Highly reported miRs that appear to modulate EC function either beneficially or detrimentally are presented in Table 2.
Principal endothelial microRNAs and their involvement in cell function.
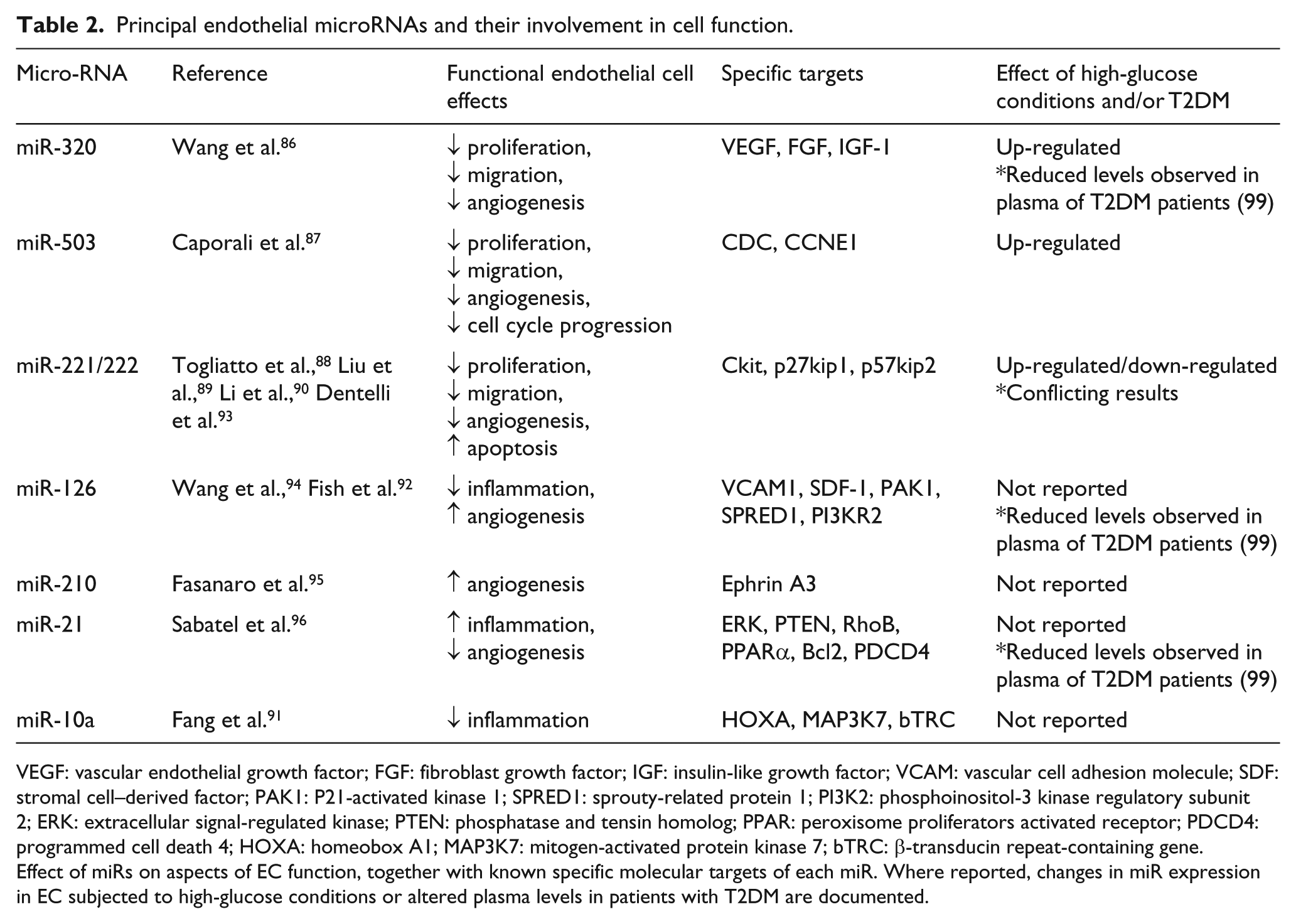
VEGF: vascular endothelial growth factor; FGF: fibroblast growth factor; IGF: insulin-like growth factor; VCAM: vascular cell adhesion molecule; SDF: stromal cell–derived factor; PAK1: P21-activated kinase 1; SPRED1: sprouty-related protein 1; PI3K2: phosphoinositol-3 kinase regulatory subunit 2; ERK: extracellular signal-regulated kinase; PTEN: phosphatase and tensin homolog; PPAR: peroxisome proliferators activated receptor; PDCD4: programmed cell death 4; HOXA: homeobox A1; MAP3K7: mitogen-activated protein kinase 7; bTRC: β-transducin repeat-containing gene.
Effect of miRs on aspects of EC function, together with known specific molecular targets of each miR. Where reported, changes in miR expression in EC subjected to high-glucose conditions or altered plasma levels in patients with T2DM are documented.
In summary, while the study of epigenetic mechanisms associated with diabetes and its complications has gained momentum in recent years, there is still much to be learned. Moreover, the complexity of these interactions will pose considerable challenges for their elucidation.
Current therapies for DM
Most current treatment options for reducing T2DM-related cardiovascular risk focus on controlling metabolic imbalances such as hyperglycaemia, hypertension and raised levels of FFA. While insulin is the obvious treatment of choice in T1DM, first line action for T2DM is through lifestyle changes. The large-scale Look AHEAD (Action for Health in Diabetes) trial 100 reported, however, that while weight loss in DM patients conferred multiple health benefits, cardiovascular risk remained unchanged. 101 Oral hypoglycaemic drugs aim to correct the pathophysiological features that lead to T2DM generally by targeting the key characteristic of insulin resistance. Essentially, these drugs act by either increasing insulin production (insulin secretagogues such as sulphonylureas, glinides and more recently incretin mimetics) or increasing target tissue sensitivity (insulin sensitisers such as metformin and thiazolidinediones). 102 While there is genuine optimism that new oral therapies may also contribute to reducing cardiovascular risk, a systematic review showed metformin to be the only current agent with such potential. 103 While able to reduce levels of glycosylated haemoglobin and microvascular disease to some degree, oral antidiabetic drugs still fall short of ameliorating cardiovascular complications in the short or medium term, and importantly have been implicated in increased mortality from hypoglycaemia 104 or myocardial infarction. 105 Nevertheless, trials have highlighted the importance of early glycaemic intervention to reduce vascular complications, likely due to the effects of metabolic memory.
Approach to future therapies
This review has highlighted the critical involvement of endothelial dysfunction in the pathogenesis of vascular disease in DM, in which diabetic mediators inflict a pro-atherogenic, inflammatory phenotype, often with limited or delayed reversibility. Owing to the silent nature of insulin resistance and hence an inevitable problem in rapid diagnosis, early intervention is difficult. Manipulation or inhibition of some of the described mechanisms may provide novel avenues for therapeutic intervention. However, the transition from this experimental stage through to production of safe and effective drugs is not easy, and requires results from valid and reliable large-scale clinical trials. Such clinical trials have already revealed disappointing outcomes regarding the use of anti-oxidants in alleviating oxidative stress;59,60 however, they should not deter further anti-oxidant therapy trials. It is likely that a ‘prevention rather than cure’ approach is preferable in the management of ROS, with the enzymatic sources of superoxide anion a promising target for future therapeutics. 16 Other mechanisms meriting further investigation include PKC inhibition 65 and the previously mentioned use of salicylate anti-inflammatory drugs. 45
Rapidly accumulating evidence supports the notion that miRs are cell-type specific and differentially expressed, and may actually play a causative role in diabetes and its related vascular complications. 97 Recent evidence suggests that miRs can be communicated between cells and specifically, EC-derived plasma miRs are reportedly able to regulate the biological functions of not only other EC, but also vascular SMC and leukocytes (reviewed in Yamakuchi 106 ). In future, understanding and exploiting these mechanisms has potential to be translated into defining clinical biomarkers and elucidating specific molecular targets for therapeutics. In the longer term, these may be of value in reversing the devastating outcomes attributed to endothelial dysfunction in diabetes by erasing persistent metabolic memory by ‘re-setting’ the epigenetic cell signature.
Conclusion
Recent advances in knowledge surrounding metabolic memory and the role of epigenetics have provided exciting new avenues for future research. However, there is still much to learn regarding their roles in diabetic vascular complications, and specifically on the EC. To date, in vitro studies of metabolic memory in cultured EC have largely focused on changes in gene expression in response to hyperglycaemia. 80 However, this review has highlighted the contribution of a complex array of factors other than glucose in the pathogenesis of endothelial dysfunction. For this reason, it is apparent that identifying the presence of a diabetic metabolic memory might be most accurately observed in EC derived from human diabetic vessels. In such a way, we have previously identified inherent functional differences between vascular SMC cultured from patients with and without T2DM, 107 such data offering an explanation for increased venous bypass graft stenosis after revascularisation surgery in these patients. Identification of any sustained functional differences in T2DM-EC may provide further justification for the aforementioned graft failure. Such information would potentially create a foundation, upon which therapeutics to ameliorate endothelial dysfunction could be developed.
Footnotes
Declaration of conflicting interests
The authors declare no conflict of interest.
Funding
Anna C Roberts undertook this work as part of an Intercalated Bachelor of Science degree in Cardiovascular Medicine, funded by the Alumni Footsteps Fund at the University of Leeds.