
Abstract
Numerous epidemiological studies have consistently demonstrated the strong association between type 2 diabetes mellitus (T2DM) and an increased risk to develop cardiovascular disease. The pathogenesis of T2DM and its complications are characterized by pancreatic, adipose tissue and vascular inflammation. CD40 and CD40L, members of the tumour necrosis factor (receptor) TNF(R) family, are well known for their role in immunity and inflammation. Here we give an overview on the role of CD40–CD40L interactions in the pathogenesis of T2DM with a special focus on pancreatic, adipose tissue and vascular inflammation. In addition, we explore the role of soluble CD40L (sCD40L) as a potential biomarker for the development of cardiovascular disease in T2DM subjects. Finally, the therapeutic potential of CD40–CD40L inhibition in T2DM is highlighted.
Introduction
Numerous epidemiological studies have consistently demonstrated the strong association between type 2 diabetes mellitus (T2DM) and cardiovascular disease (CVD). T2DM patients have a 200% increased risk of cardiovascular death compared to the non-diabetic population, and vascular complications such as coronary artery disease and peripheral artery disease are the major cause of morbidity and mortality in T2DM.1,2 The increasing global prevalence of diabetes makes the prevention and treatment of its vascular complications a public health priority. 3 Hence, identification of those individuals at risk for vascular complications within the heterogeneous population of T2DM patients is essential to prevent and reduce vascular morbidity and mortality. 4
In T2DM, two types of vascular diseases are distinguished: microvascular diseases such as retinopathy, neuropathy and nephropathy, and macrovascular disease such as coronary artery disease, cerebrovascular disease and peripheral arterial disease. 5 The pathophysiology of T2DM that eventually results in the development of microvascular and/or macrovascular complications is complex and remains incompletely understood.
The key pathophysiological characteristics of T2DM include insulin resistance (IR), glucose intolerance, hyperinsulinemia, hyperglycaemia and diabetic dyslipidemia. 5 In addition, T2DM is commonly complicated by the presence of co-morbidities including obesity, metabolic syndrome and hypertension, among others. Together these factors promote a chronic, systemic, low-grade inflammation, eventually resulting in the progression of T2DM and the development of vascular disease. 5 Increasing evidence suggest that the tumour necrosis factor (TNF) (receptor) (TNF(R)) family members CD40 and CD40L contribute to the T2DM-associated inflammation and subsequent development of vascular complications.
CD40–CD40L
The co-stimulatory molecule CD40 and its ligand CD40L (CD154, GP139) are expressed on not only immune cells, including B cells, T cells, dendritic cells and monocytes, but also on non-immune cells, including platelets, endothelial cells, fibroblasts, pancreatic islet β-cells and pancreatic ductal cells.6–9 Besides a membrane-bound form (mCD40L), CD40L also exists as a soluble molecule; soluble CD40L (sCD40L), mainly derived from activated platelets and T cells. 10 Although the exact biological function of sCD40L remains elusive, two-nested case-control studies and two cohort studies, with a total number of 2660 participants free from CVD demonstrated that increased levels of sCD40L were associated with myocardial infarction and cardiovascular death. The adjusted relative risk estimates for sCD40L varied between 1.9 and 2.8. 11 Although sCD40L can be used as a biomarker, sCD40L by itself also has biological activity. For example, Hristov et al. 12 demonstrated that sCD40L reduced the viability and proliferative capacity of endothelial progenitors, thereby possibly contributing to endothelial dysfunction. In addition, sCD40L has been shown to promote platelet activation and proliferation and migration of vascular smooth muscle cells.13,14
CD40–CD40L interactions are required for many immune processes, for example, chemokine and cytokine production, B cell activation, co-stimulation, immunoglobulin isotype switching and memory cell formation.6,10 In addition, CD40–CD40L signalling is involved in the pathophysiology of numerous inflammatory diseases, including atherosclerosis, inflammatory bowel disease, systemic lupus erythematosus (SLE), rheumatoid arthritis, type 1 diabetes mellitus and allograft rejection.15–18 For example, in atherosclerosis CD40–CD40L interactions critically contribute to the development of atherosclerotic plaques in the vessel wall. Inhibition of the CD40–CD40L axis in murine models of atherosclerosis results in the formation of smaller atherosclerotic plaques, characterized by a clinically favourable stable phenotype, which is low in inflammatory cell numbers and high in fibrosis.16,17 Until now, inhibition of CD40–CD40L is one of the most powerful plaque-stabilizing strategies in laboratory settings.
In recent years, experimental and clinical studies have demonstrated the involvement of the CD40–CD40L axis in the development and progression of T2DM and its vascular complications. Plasma levels of sCD40L are elevated in sub-optimally treated, hyperglycaemic and dyslipidemic T2DM patients and sCD40L levels may also predict the occurrence of cardiovascular events in these patients.19–21 Experimental studies suggest that inhibition of CD40–CD40L may reduce the systemic inflammatory response responsible for the progression of T2DM and the development of CVD, as discussed below.8,9,22
CD40–CD40L interactions in the pathogenesis of pancreatic inflammation in T2DM
Chronic, low-grade inflammation is a hallmark of T2DM and is manifest in the pancreas, adipose tissue, liver, vasculature and in the circulation. 5 The inflammatory responses are characterized by increased leukocyte numbers and elevated expression of pro-inflammatory cytokines, such as interleukin (IL)-1β and IL-6, 23 which are induced and maintained by oxidative stress, lipotoxicity, glucotoxicity and ectopic lipid deposition. 5
The co-stimulatory molecule CD40 is expressed on both human and mouse pancreatic islet β-cells and pancreatic duct cells, but not on α-cells. 9 The expression of CD40 on pancreatic islet and ductal cells is increased upon exposure to pro-inflammatory cytokines, including TNF-α, IL-1β and interferon (IFN)-γ, all abundantly present in the diabetic pancreas.8,9 Whereas membrane-bound CD40L is expressed on immune cells that infiltrate the diabetic pancreas, sCD40L is cleaved from the surface of activated platelets by the proteinase ADAM 10. 10 Although the biological effects of sCD40L-mediated signalling in T2DM remain largely unknown, binding of membrane-bound CD40L to CD40 on β-cells results in the activation of nuclear factor kappa B (NFκB), and subsequently induces the expression of cytokines, including IL-6, IL-8 and chemokines, such as monocyte chemoattractant protein (MCP)-1 and macrophage inflammatory protein (MIP)-1β.8,9 This further enhances pancreatic inflammation and impairs β-cell insulin release or production. Although in vitro experiments suggested that insulin metabolism is not affected by CD40L signalling, Poggi et al. 22 recently demonstrated that obese CD40L−/− mice have preserved insulin sensitivity and low plasma insulin levels compared to obese CD40L+/+ mice. 8 However, CD40L−/− mice were not more glucose tolerant, probably as a result of decreased plasma insulin levels. 22 The effects reported in CD40L−/− mice could be mimicked by antibody-mediated inhibition of CD40L in obese CD40L+/+ mice. 22 Interestingly, the improvement of insulin sensitivity and decreased plasma insulin concentrations in anti-CD40L antibody treated mice was independent from body weight, 22 suggesting that CD40L may have direct effects on insulin metabolism, at least in a mouse model of diet-induced obesity.
CD40–CD40L interactions may thus induce or exacerbate pancreatic inflammation in T2DM, thereby indirectly impairing insulin metabolism, and initial studies suggest a direct role for CD40–CD40L in insulin production or release. Future studies are required to explore the full effects of the CD40-signalling pathway on insulin metabolism.
CD40–CD40L promotes adipose tissue inflammation in T2DM
Systemic inflammation is a key feature of T2DM and its co-morbidities such as obesity and the metabolic syndrome.24,25 Systemic inflammation aggravates the progression of T2DM and the development of vascular complications. In recent years, it has been demonstrated that adipose tissue is a major endocrine organ that contributes to the development of systemic inflammation in various inflammatory diseases, such as obesity, atherosclerosis and T2DM.25,26
CD40 messenger RNA (mRNA) levels in adipose tissue and plasma levels of sCD40L positively correlate with body mass index, and CD40 is expressed on adipocytes and stromal adipose tissue, including immune cells. 27 CD40L signalling in adipose tissue increases the expression of pro-inflammatory mediators, including TNF-α, IL-6 and MCP-1. 27 Furthermore, CD40L stimulation enhances lipid droplet accumulation and adipogenesis in 3T3-L1 cells. 28 Interestingly, CD40L exposure reduces insulin-mediated glucose uptake by adipocytes, as a result of reduced expression of insulin receptor substrate (IRS-1) and glucose transporter type-4 (GLUT-4), whereas IRS-2 and GLUT-1 levels remain unaffected.22,28 These findings could be reproduced by co-culture of adipocytes and T cells, the main source of membrane-bound CD40L. 22 Thus, in vitro experiments suggest that CD40L+ T cells may directly induce adipocyte inflammation and impair adipose tissue insulin sensitivity, thereby contributing to systemic inflammation and IR.
IR results in increased plasma glucose levels and compensatory hyperinsulinemia. 29 Both hyperglycaemia and chronic hyperinsulinemia increase the expression of membrane-bound CD40L on circulating platelets, platelet CD40L subsequently promotes the formation of platelet-leukocyte aggregates and leukocyte-endothelium interactions, thereby enhancing vascular inflammation.30,31 In contrast to chronic hyperinsulinemia in T2DM, short-term hyperinsulinemia decreased plasma sCD40L levels. 32 Although the underlying mechanisms were not elucidated, one can speculate that short-term hyperinsulinemia mimics the physiological response to transient fluctuations in glucose metabolism and does not reflect inflammatory conditions, whereas chronic hyperinsulinemia reflects a pathological condition. 32
In conclusion, CD40–CD40L signalling plays a major role in adipose tissue inflammation, adipogenesis and IR. Together these factors promote systemic inflammation, thereby stimulating the progression of T2DM and its complications. However, current evidence is mainly based on correlation studies and in vitro studies, emphasizing the need for additional in vivo studies to unravel the exact role of CD40–CD40L in adipose tissue inflammation in T2DM.
The key role of CD40–CD40L in vascular inflammation
Atherosclerosis, the pathological substrate of macrovascular disease in T2DM, is the result of chronic inflammation of the large- and mid-sized arteries. 33 The immune system plays an important role in the development of atherosclerotic plaques. Plaque rupture subsequently results in thrombosis and vascular occlusion, thereby causing clinical symptoms, such as peripheral occlusive artery disease and myocardial infarction. 33 In the last decade, we and others have demonstrated a critical role of the CD40–CD40L dyad in the development of atherosclerosis.6,15–17 Both genetic and pharmacologic inhibition of CD40(L) signalling reduced plaque size and induced a plaque phenotype characterized by high levels of collagen and low numbers of inflammatory cells.15,16,34 Interestingly, even when anti-CD40L treatment was started after plaques had developed, these plaques transformed into this beneficial phenotype.
Both CD40 and CD40L are expressed on immune cells and non-immune cells present on cells in the atherosclerotic plaque, as discussed above. In initial atherosclerotic plaques CD40–CD40L interactions promote leukocyte recruitment by at least three mechanisms. First, activation of CD40 on endothelial cells results in the expression of adhesion molecules, including vascular cell adhesion molecule (VCAM)-1, intercellular cell adhesion molecule (ICAM)-1 and E-selectin. 35 Second, macrophages present in initial plaques secrete pro-inflammatory chemokines upon CD40L exposure such as MCP-1, MIP-1α, MIP-1β and Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES).36,37 Third, platelet CD40L contributes to the formation of platelet–leukocyte aggregates and promotes leukocyte adhesion to the activated endothelium. 31
In advanced plaques, CD40–CD40L interactions further aggravate inflammation by stimulating macrophage cytokine production (IL-1β, IL-2, IL-6 and TNFα), and CD40-induced production of matrix metalloproteinases contributes to destabilization of the atherosclerotic plaque, thereby promoting plaque rupture and subsequent arterial occlusion.36–38
Unfortunately, antibody-mediated blockage of CD40L was complicated by thromboembolism when applied to patients, as a result of disrupted CD40L-αIIbβ2 interactions in arterial thrombi. 39 Therefore, clinical trials using the anti-CD40L antibody were stopped. Furthermore, investigation of CD40L-CD40 induced signalling pathways is required to identify therapeutic targets without these side effects. Our group recently evaluated the role of leukocyte-specific CD40L-CD40-TNF receptor-associated factor (TRAF) signalling in atherosclerosis. Since CD40 has no intrinsic signalling ability, it requires TRAFs to elicit intracellular signalling. The cytoplasmatic domain of CD40 contains a proximal binding site for TRAF6, and two distal sites that bind TRAF1, TRAF2, TRAF3 and, indirectly, TRAF5. To evaluate the contribution of leukocyte-specific CD40L-CD40-TRAF2/3/5 and CD40L-CD40-TRAF6 signalling to atherosclerosis, we generated ApoE−/−CD40−/− mice expressing a chimeric CD40 transgene with mutations at the TRAF6 and TRAF2/3/5 binding site under the control of class II major histocompatibility complex (MHCII) promotor. 15 Deficiency of CD40-TRAF2/3/5 signalling did not affect atherogenesis. 15 Interestingly, deficiency in CD40-TRAF6 interactions abrogated atherosclerosis as a result of reduced blood counts of Ly6Chigh monocytes, impaired recruitment of Ly6C+ monocytes to the arterial wall and polarization of macrophages towards the anti-inflammatory M2 phenotype. 15 Hence, inhibition of CD40L-CD40-TRAF6 signalling may become a promising therapeutic target for atherosclerosis.
Do sCD40L levels reflect the inflammatory status in T2DM patients?
Glycated haemoglobin A1c (HbA1c) is used as a marker for glycaemic control of diabetes. 35 Current guidelines state that the treatment of T2DM should aim to reduce HbA1c levels to 7% of total haemoglobin, or even lower. 40 However, an increasing amount of evidence indicates that HbA1c may not adequately predict the development of microvascular and macrovascular complications.2,41–45 Thus, additional biomarkers, that reflect systemic inflammation, may be required to identify the T2DM patients who are at risk to develop macrovascular complications.
sCD40L levels may predict the risk of major cardiovascular events, including acute myocardial infarction, sudden cardiac death and recurrent angina in patients suffering from coronary artery disease.46–48 For example, Lobbes et al. 11 demonstrated that sCD40L levels predict the occurrence of myocardial infarction and cardiovascular death in a population free from CVD, as discussed above. Interestingly, T2DM is independently associated with elevated sCD40L levels in patients suffering from acute coronary syndromes and myocardial infarction.49,50 In T1DM, sCD40L levels have been shown to correlate with endothelial dysfunction, monocyte activation and increased numbers of platelet-leukocyte aggregates.50,51 Although elevated levels of sCD40L were associated with carotid atherosclerosis in T1DM, elevated levels did not predict cardiovascular mortality.52,53
As discussed above, IR, hyperinsulinemia, hyperglycaemia and obesity are all associated with increased sCD40L levels. 54 Correlative studies have demonstrated that treatment of these factors may reduce plasma levels of sCD40L. For example, thiazolidinediones reduced sCD40L levels by 27% in recent onset T2DM, while in long-standing disease, sCD40L was reduced by 29% in patients without macrovascular complications and by 34% in patients suffering from these complications. 55 Furthermore, treatment of diabetic dyslipidemia using atorvastatin reduced sCD40L levels in T2DM patients who suffered from coronary artery disease. 56 However, conflicting study outcomes are reported21,57–60 in Table 1, and we suggest that this is the result of heterogeneity in study protocols. At least three factors may contribute to this heterogeneity. As mentioned, activated platelets are the main source of sCD40L, hence the levels of the protein differ between plasma and serum samples. The highest levels have been reported in serum samples, and sCD40L levels in clotting plasma increased with time.61,62 Since low temperatures inhibit the increase in serum samples, it is likely that the increase is the result of ex vivo release of sCD40L from activated platelets.61–63 In addition, Dominguez-Rodriguez et al. 64 observed a circadian rhythm in sCD40L levels in patients suffering from myocardial infarction. Samples drawn at 9 p.m. contained 41.5% higher sCD40L levels than samples obtained at 2 a.m. Although the exact mechanisms were not investigated, it was hypothesized that circadian differences in proteinase levels may be involved. 64 Finally, the use of anti-platelet drugs, such as Cyclooxygenase inhibitors or adenosine diphosphate receptor inhibitors, may affect the release of sCD40L from platelets. 65 Hence, the use of this type of drugs should be carefully monitored in clinical studies.
Conflicting results regarding the effects of anti-hyperglycaemic and anti-hyperlipidemic drugs on serum or plasma sCD40L levels in T2DM subjects have been reported. The heterogeneity may be the result of different sCD40L levels in plasma and serum, a circadian rhythm in sCD40L release, and the use of anti-platelet drugs.
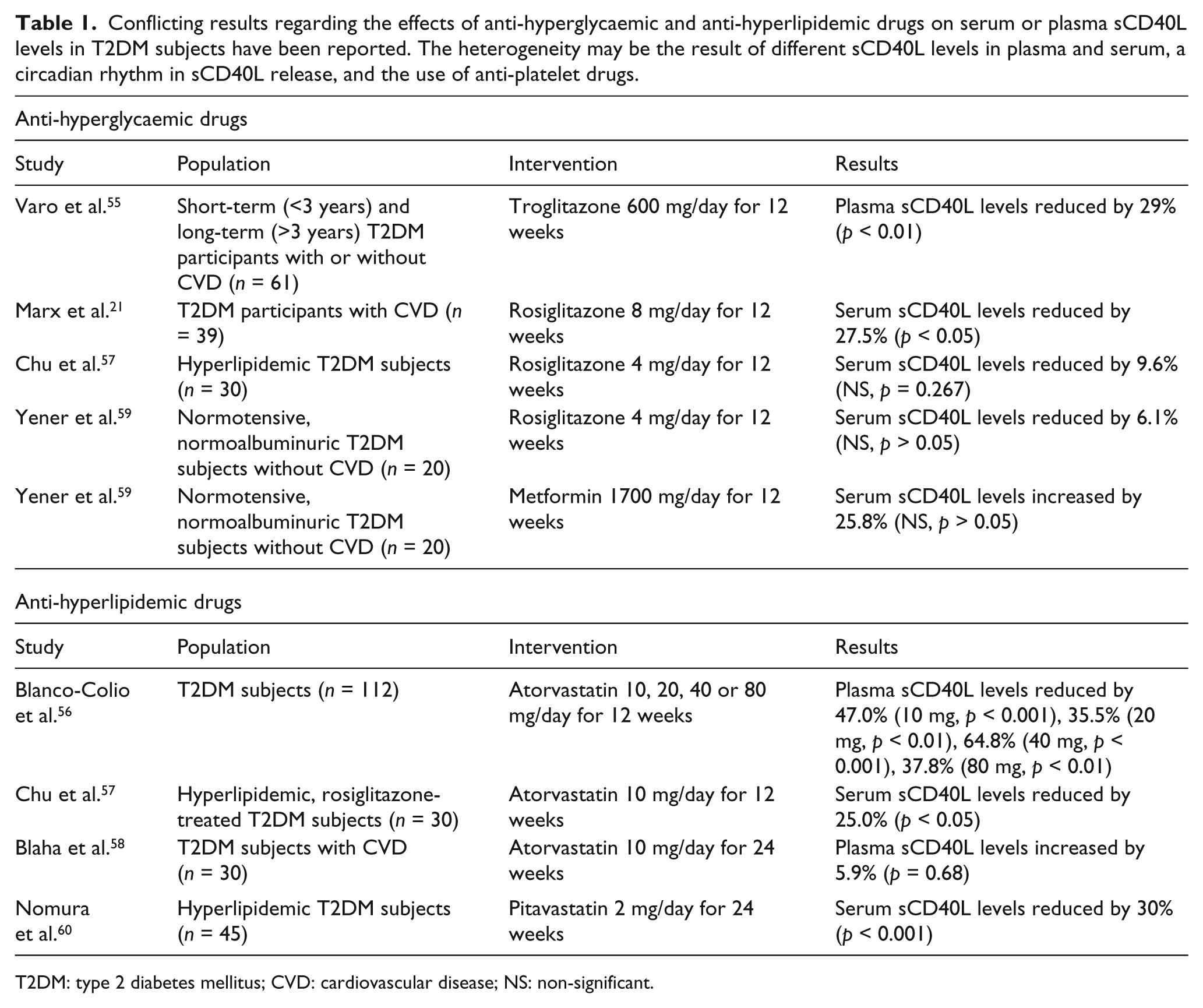
T2DM: type 2 diabetes mellitus; CVD: cardiovascular disease; NS: non-significant.
Thus, both experimental and clinical studies suggest that sCD40L may reflect the inflammatory status of T2DM patients, and may be used as a biomarker to identify those who are at risk to develop CVD. However, standardization of study protocols is of great importance to evaluate the full potential of sCD40L as a novel biomarker.
CD40–CD40L as a therapeutic target in T2DM
Commonly used anti-diabetic drugs may reduce inflammation in T2DM. For example, metformin and thiazolidinediones have been shown to reduce C-reactive protein (CRP) levels. 66 These effects are not merely due to glucose lowering, because the reductions in inflammatory markers are greater than the reduction seen after similar glucose lowering induced by other anti-hyperglycaemic approaches.66,67 Nevertheless, these drugs do not prevent the occurrence of CVD in a major number of T2DM patients, emphasizing the need for additional anti-inflammatory therapeutic strategies. For example, the blockade of the pro-inflammatory cytokine IL-1β improved endocrine pancreas function, and reduced hyperglycaemia and systemic inflammation in T2DM patients.68,69
The therapeutic potential of CD40 and CD40L has been investigated in multiple inflammatory diseases, including atherosclerosis, inflammatory bowel disease, psoriasis, SLE, rheumatoid arthritis, allograft rejection and type I diabetes. 18 The anti-CD40 monoclonal antibody (mAb) ch5D12 reduced disease severity in Crohn’s disease, and Ruplizimab, an anti-CD40L mAb, was successfully used in the treatment of SLE and inflammatory bowel disease. 10 In addition, Poggi et al. 22 demonstrated that anti-CD40L mAb (MR-1) treatment improved insulin sensitivity in a mouse model of diet-induced obesity. Thus, specific inhibition of CD40L-CD40 signalling may become a promising strategy to improve adipose tissue and vascular inflammation, thereby reducing the occurrence of vascular complications of T2DM.
Concluding remarks and future directions
The CD40–CD40L dyad has a complex and not fully elucidated role in the pathogenesis of T2DM. However, both experimental and clinical studies show that CD40–CD40L interactions promote pancreatic, adipose tissue and vascular inflammation, and induce a chronic, systemic inflammatory response that eventually results in the formation of atherosclerosis (Figure 1).
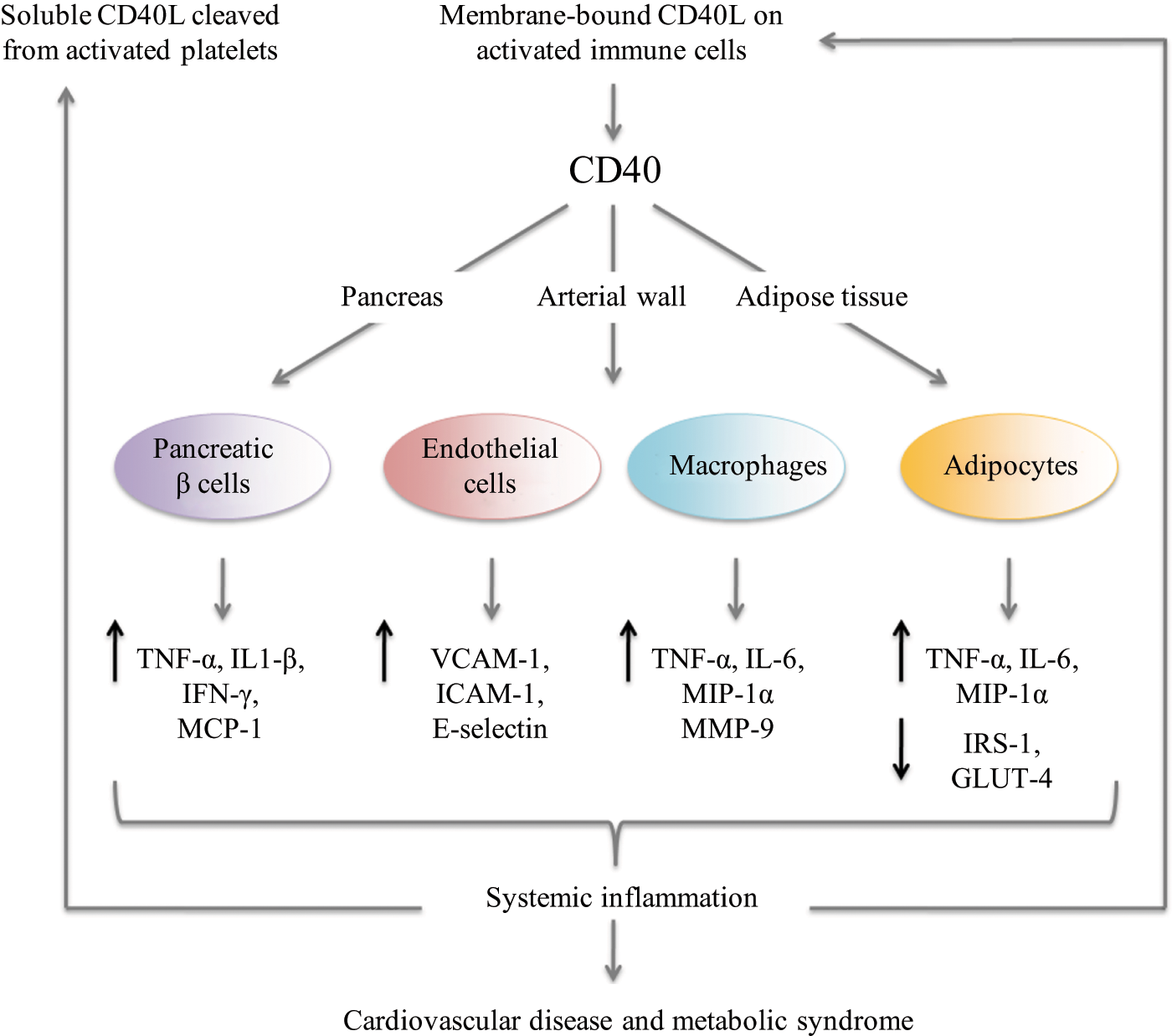
CD40L binding to CD40 results in the up-regulation of pro-inflammatory cytokines and chemokines by pancreatic islet β cells, macrophages and adipocytes. The expression of adhesion molecules on endothelial cells is increased. Insulin-mediated glucose uptake is impaired in adipocytes as a result of decreased IRS-1 and GLUT-4 expression. Together, CD40–CD40L interactions induce a systemic inflammatory response that promotes not only the development of CVD and the metabolic syndrome but also further increases the expression on CD40L and elevates the levels of sCD40L.
Numerous clinical studies have demonstrated that sCD40L levels are elevated in patients suffering from diabetes and its complications. Although the exact inflammatory mechanisms that are responsible for this increase are not completely understood, it is clear that sCD40L reflects the inflammatory status in T2DM. Future studies should explore the potential of sCD40L as a biomarker to identify those patients who are at risk of developing vascular complications within the heterogeneous T2DM population. In order to do so, it is essential to standardize sCD40L measurement protocols. Finally, elucidation of CD40–CD40L signalling pathways involved in diabetic inflammation may identify novel therapeutic targets for T2DM and its complications.
Key messages
CD40–CD40L interactions promote pancreatic, adipose tissue and vascular inflammation in T2DM.
sCD40L may become a biomarker for the development of CVD in patients with T2DM.
Specific inhibition of CD40–CD40L signalling may reduce the occurrence of vascular complications of T2DM.
Footnotes
Funding
This study was supported by the Humboldt Foundation (Sofja Kovalevskaja grant to E.L.), the Netherlands Organization for Scientific Research (VIDI grant to E.L.), the Netherlands Heart Foundation (Dr E. Dekker MD-grant to T.S. + Dr E. Dekker Established Investigator grant to E.L.).