
Abstract
Mainstream and sidestream tobacco smoke extracts have been shown to increase platelet activation directly. Furthermore, advanced glycation end products, which are present in the diabetic vasculature, have also been shown to enhance platelet activity. However, the combined effects of these two risk factors on platelet functions remain unclear. Platelets were exposed to tobacco extracts concurrently with advanced glycation end products. Timed samples were removed to assess the extent of platelet activity. The presence of smoke extracts enhanced platelet activity as compared to control conditions, this was especially prevalent for sidestream extracts. With the addition of irreversibly glycated albumin, there was an additive effect, further enhancing platelet responses. This was at least partially regulated by α-granule release and CD41 expression. The combination of cardiovascular risk factors can significantly enhance platelet activation and aggregation, and therefore it is possible to accelerate cardiovascular diseases through the interactions of multiple cardiovascular risk factors.
Keywords
Introduction
It has been well established that tobacco smoke is a primary risk factor for cardiovascular disease development, including atherosclerosis and peripheral artery disease. 1 Diabetes is also considered as a cardiovascular risk factor, primarily due to the actions of advanced glycation end products. 2 However, there has been little work on the combined interactions of these two risk factors to determine if cardiovascular disease progression accelerates when a diabetic smokes or is exposed to the more potent sidestream tobacco smoke. Both tobacco smoke (mainstream and sidestream) and glycated proteins have been shown to alter the hemostatic response through an enhancement of platelet activation, aggregation and adhesion.3–5 Enhancement of these functions has also been shown to trigger cardiovascular diseases and promote oxidative stress production or endothelial cell nitric oxide synthase (NOS) activation, which can further perpetuate cardiovascular disease progression.6,7 A compounding factor to tobacco smoke induced cardiovascular disease development is that nicotine, the major psychoactive ingredient in cigarettes, can inhibit many hemostatic functions, while having many deleterious effects in other systems.4,8,9
Advanced glycation end products are formed when molecules are exposed to sugars for a long period of time. 10 During diabetes, plasma proteins are especially susceptible to glycation due to the low turnover rate of the proteins and the restriction of glucose to the vascular space. Recent work from our group has suggested that platelets and endothelial cells respond differently to albumin glycated to various extents.5,11 In general, early, potentially reversibly, glycation is not associated with pathological cellular changes whereas, late, potentially irreversibly, glycation is associated with pathological cellular changes. There have been few studies which reported how tobacco smoke and nicotine interact with glycated proteins to alter platelet functions and these reports suggest that combining cardiovascular risks accelerates cardiovascular disease progression.12,13 It is crucial to elucidate how cardiovascular risk factors interact to mediate disease progression.
Many have shown that enhanced thrombin generation and platelet aggregation are salient steps during cardiovascular disease progression. During and after smoking, platelet-induced thrombin generation is elevated and this appears to be independent of nicotine. 14 Platelet aggregation responses are also enhanced during and after smoking and this is mediated through an activation of adhesion receptors. 15 Furthermore, diabetics have been shown to have an elevated platelet activity.16,17 Therefore, it is likely that combinations of advanced glycation end products and tobacco smoke can lead to a sustained enhancement of the hemostatic system.
In the present study, we measured the thrombogenic and aggregation potential of platelets exposed to mainstream or sidestream tobacco smoke extracts concurrently with early or late advanced glycation end products. We also investigated the role of nicotine on these properties. We hypothesised that the combination of multiple cardiovascular risk factors would accelerate platelet activation/aggregation mediated through α-granule release and CD41 expression. We also hypothesised that nicotine would not play a role in this heightened activation potential.
Methods
Smoke extracts/nicotine
Tobacco smoke extracts were made using the method reported by Rubenstein et al. with minor modifications. 4 One high-tar cigarette (16 mg tar and 1.2 mg nicotine, Philip Morris) was used to make mainstream, mimicking the smoker, and sidestream, mimicking second hand smoke, extracts. For all preparations, puffing was mimicked by alternating the airflow rate every 30 seconds from 200 mL/min (25 sec) to 500 mL/min (5 sec). This procedure was followed until the cigarette burned to within 2mm of the filter (~5 min). Smoke was bubbled through a 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) buffered saline (130 mM NaCl, 20 mM HEPES, pH 7.4) via a step-down manifold to 6–0.9 mm internal diameter PTFE tubing (Small Parts Inc.). Standard extracts were made from 1 cigarette into 100 mL of buffer. Samples were aliquoted and stored at -20 °C until use. Tobacco smoke extracts were added to platelet samples at a concentration of 1 cigarette/5L, which is the approximate blood concentration, assuming that all smoke enters the blood stream as either mainstream (smoker) or sidestream (non-smoker) after being exposed to one cigarette. (-)-Nicotine (Sigma Aldrich) was added directly to platelet preparations at 50 nM, which is a concentration within the range expected in the plasma immediately after smoking a cigarette. 18
Advanced glycation end products
Bovine serum albumin was glycated as previously reported by us without modifications. 5 The batch of glycated albumin that was used in this study was chemically modified with an average of 6 glucose molecules after 2 weeks of glycation, 25 glucose molecules after 6 weeks of glycation and 30 glucose molecules after 8 weeks of glycation. Samples were aliquoted and stored at -4 °C until use, as previously described. 2 mg/mL glycated albumin (denoted as AGE) or non-glycated albumin (denoted as BSA) was added to platelet samples for up to 4 hours. All experiments were compared to a sample with no additional albumin and no tobacco smoke, denoted as ‘Negative Control’.
Platelets
Fresh platelet-rich plasma pheresis packs (anti-coagulated with 0.32% sodium citrate) were obtained from the Oklahoma Blood Institute (Oklahoma City, OK). For platelet activation measurements (prothrombinase assay) and flow cytometry measurements, washed platelets were obtained by centrifugation of platelet-rich plasma for 9 min at 3000 rpm (1100 × g). The platelet pellet was re-suspended in a platelet buffer as previously reported. 5 Platelets that were not in use were maintained at room temperature under gentle agitation.
Prothrombinase assay
The prothrombinase assay quantifies the rate of thrombin generation and is dependent on the amount of factor Va released from the platelet α-granule and the amount of phosphatidylserine expressed on the platelet outer leaflet. 19 Prior to the start of this assay, prothrombin is acetylated, which prevents the positive feedback of thrombin on platelets. This provides a 1:1 relationship between platelet activity and thrombin generation, which is quantified with Chromozym-Th (Roche Applied Sciences); a chromogenic substrate for thrombin. For complete details on this assay please refer to Rubenstein & Yin. 5 Paired platelet samples were maintained with their particular additive for up to 2.5 hours under static conditions.
Optical platelet aggregation
Platelet-rich plasma (diluted to 250,000/µL with autologous platelet-poor plasma) was warmed to 37 °C in a microcuvette, stirred at 1000 rpm in an aggregometer (Chrono-Log Model 490-2D, Havertown, PA). 1 minute after placing the cuvette in the aggregometer, aggregation was initiated through the addition of 20 µM thrombin receptor agonist peptide 6 (TRAP6). Light transmission, compared against autologous platelet-poor plasma, was recorded until clot stabilisation. Paired platelet samples were maintained with their particular additive for up to 4 hours. To confirm the role of GPIIb/IIIa, some platelet samples were incubated with the particular tobacco smoke extracts and glycated albumin additives along with 50 ng/mL tirofiban (Aggrastat from Sigma). Others have shown that 50 ng/mL tirofiban minimally inhibits the normal aggregation response to 20 µM TRAP6. 20 Therefore, the role of GPIIb/IIIa in the aggregation changes observed can be quantified with tirofiban addition.
Flow cytometry
Timed samples of washed platelets were assayed for the surface expression of CD41 (GPIIb) and CD62P (P-selectin) to determine a possible mechanism for altered aggregation and thrombin generation, respectively. PE-anti-CD41 (1:50, Ancell) and FITC-anti-CD62P (1:50, Ancell) were incubated with platelet samples for 30min at room temperature in the dark. Platelets were then diluted 1:5 in platelet buffer and immediately analysed on a flow cytometer (Accuri C6) for receptor expression data. All mean fluorescence were normalised to MOPC (negative control) and sonicated (10 sec) cells (positive control).
Statistical analysis
The platelet activity state (PAS) is quantified as the concentration of thrombin generated at each time point, normalised to the amount of thrombin generated for the negative control sample at time zero. PAS data was regressed over the 2.5 hour time course for each independent experiment and the slope of these independent lines were pooled and compared (note that the regression line shown on the plots is calculated from the pooled data; this is for visual aid and is not what was used in the statistical analysis). The rate of aggregation was quantified as the slope of the light transmission over the first 60sec after aggregation has begun. This was normalised by the paired negative control for each individual experiment. Finally, the normalised all mean fluorescence values were pooled for independent experiments. Platelet activation data was tested for significance with a one-way ANOVA (grouped by additive). Platelet aggregation and flow cytometry data were tested for significance with a two-way ANOVA (grouped by time and additive). All statistical analyses was conducted in SAS (v9.2) with α = 0.05. Note that all experiments were conducted from different pheresis packs, to ensure independence of the platelet samples.
Results
Platelet aggregation in response to tobacco smoke extracts and glycated albumin
The effect of glycated albumin, the extent of glycation and the presence of mainstream or sidestream tobacco smoke extracts on platelet aggregation was evaluated (Figure 1(a)). As expected, there was a slight increase in the rate of aggregation with time, which is common as platelets age. The presence of mainstream smoke, regardless of the added albumin, increased platelet aggregation rate slightly over the control conditions (~5–10%). However, the combination of albumin glycated to larger extents (6 and 8 week) and mainstream smoke significantly accelerated the rate of aggregation (~25–40%). Similarly, with the addition of sidestream smoke, regardless of the albumin addition, the platelet aggregation rate significantly increased (~15%) as compared with the control conditions at all-time points (Figure 1(b)). This increase in aggregation rate was more prominent for the combined albumin glycated to large extents and sidestream extracts (~50–60%). For both tobacco smoke extracts, the interaction between albumin glycated to a lower extent (2 week) and the extract did not elicit a significant change in platelet aggregation rate as compared with the 2 week non-glycated sample.
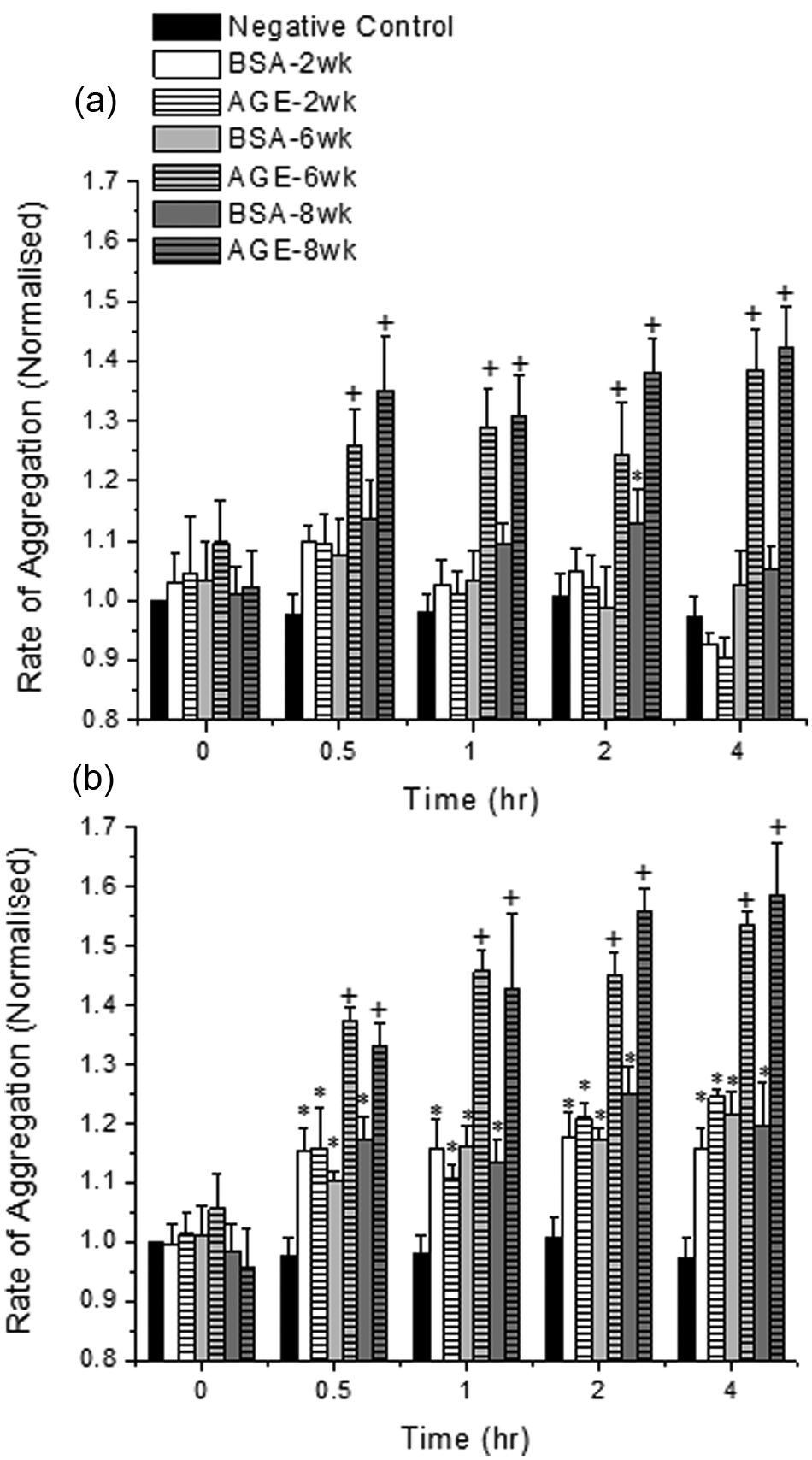
The rate of platelet aggregation in the presence of glycated (AGE) or non-glycated (BSA) albumin and mainstream tobacco smoke extracts (a) or sidestream tobacco smoke extracts (b). Platelets were kept under static conditions for these experiments. The data shown represents the mean + standard error of the mean for six independent experiments. + Differs from paired BSA (by glycation time), BSA-2 week, AGE-2 week and negative control (p < 0.05, two-way ANOVA, paired by time). *Differs from negative control only (p < 0.05, two-way ANOVA, paired by time).
Platelet aggregation in response to pure nicotine and glycated albumin
Nicotine was added to platelets that were subjected to diabetic cardiovascular conditions to determine the extent of inhibition on aggregation responses. With the addition of nicotine to any albumin sample, for over 1 hour, the rate of aggregation was not statistically different than the control conditions (Figure 2). Interestingly, at the 30 minute time point, some of the samples had a significant increase in the rate of aggregation suggesting that either: (a) the inhibitory effect of nicotine takes effect after some finite amount of time, or (b) the initial effects on aggregation due to the incubation with albumin overpowers the initial inhibitory effects of nicotine. To confirm the inhibitory effects of nicotine, we incubated our 8 week glycated or non-glycated albumin sample with sidestream tobacco smoke extracts and 50 nM nicotine. The rate of aggregation for these samples was significantly enhanced as compared to the paired negative control samples (Figure 2), but was also significantly reduced (by ~25%) as compared with the same incubations without additional nicotine (compare Figures 1(b) and 2).
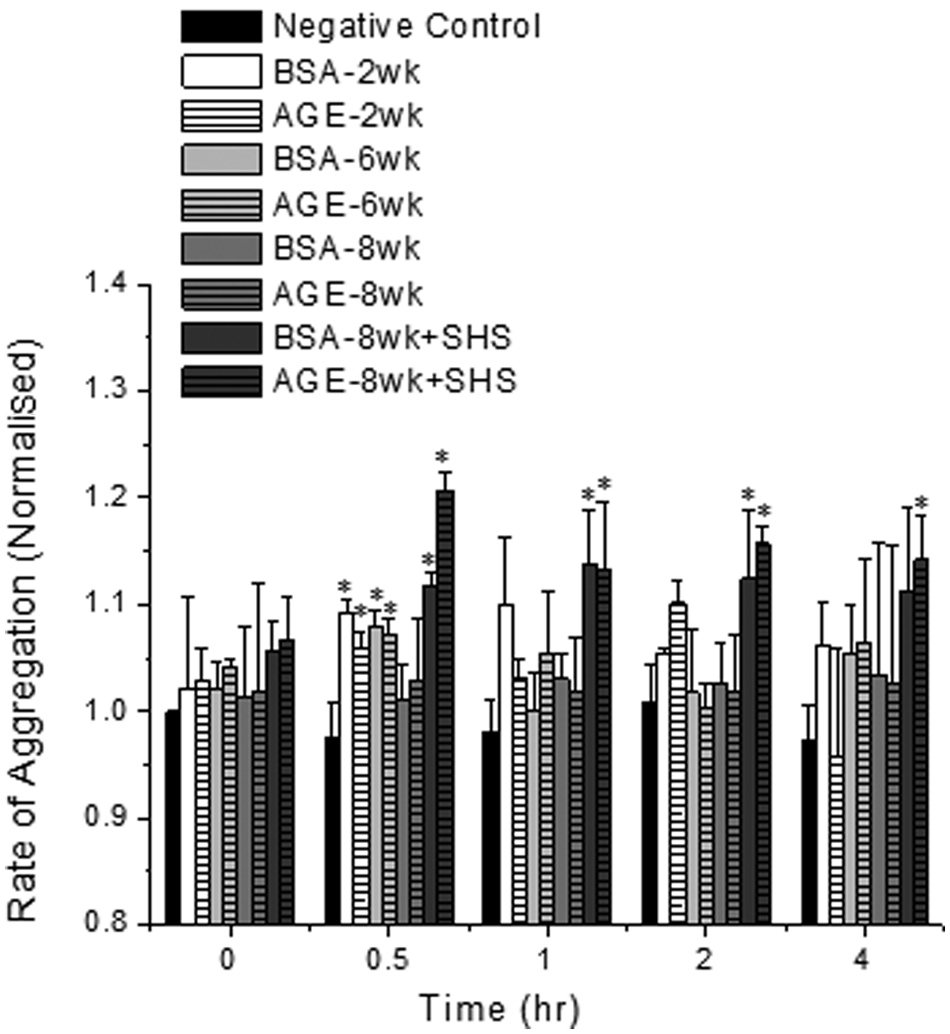
The rate of platelet aggregation in the presence of glycated (AGE) or non-glycated (BSA) albumin and 50nM nicotine. Sidestream (SHS) tobacco smoke extracts were added to some samples as noted in the legend. Platelets were kept under static conditions for these experiments. The data shown represents the mean + standard error of the mean for six independent experiments. *Differs from negative control only (p < 0.05, two-way ANOVA, paired by time).
Platelet activation in response to tobacco smoke extracts and glycated albumin
In order to determine whether or not tobacco smoke renders platelets exposed to a diabetic vasculature more susceptible to activation, we quantified the amount of thrombin generation after platelets were incubated with glycated albumin and mainstream extracts or sidestream extracts, under static conditions. With these combined incubations, the rate of platelet activation was independent of the cigarette extraction method (not shown) and was only enhanced for the irreversibly glycated albumin samples (6 week: ~60% increase and 8 week: ~120% increase) combined with mainstream (Figure 3(a)) or sidestream extracts (Figure 3(b), some data from Figure 3 is removed for ease of viewing). With the addition of nicotine, instead of tobacco smoke extracts, the rate of platelet activation was reduced back to control conditions regardless of the presence of glycated albumin or the glycation extent (not shown).
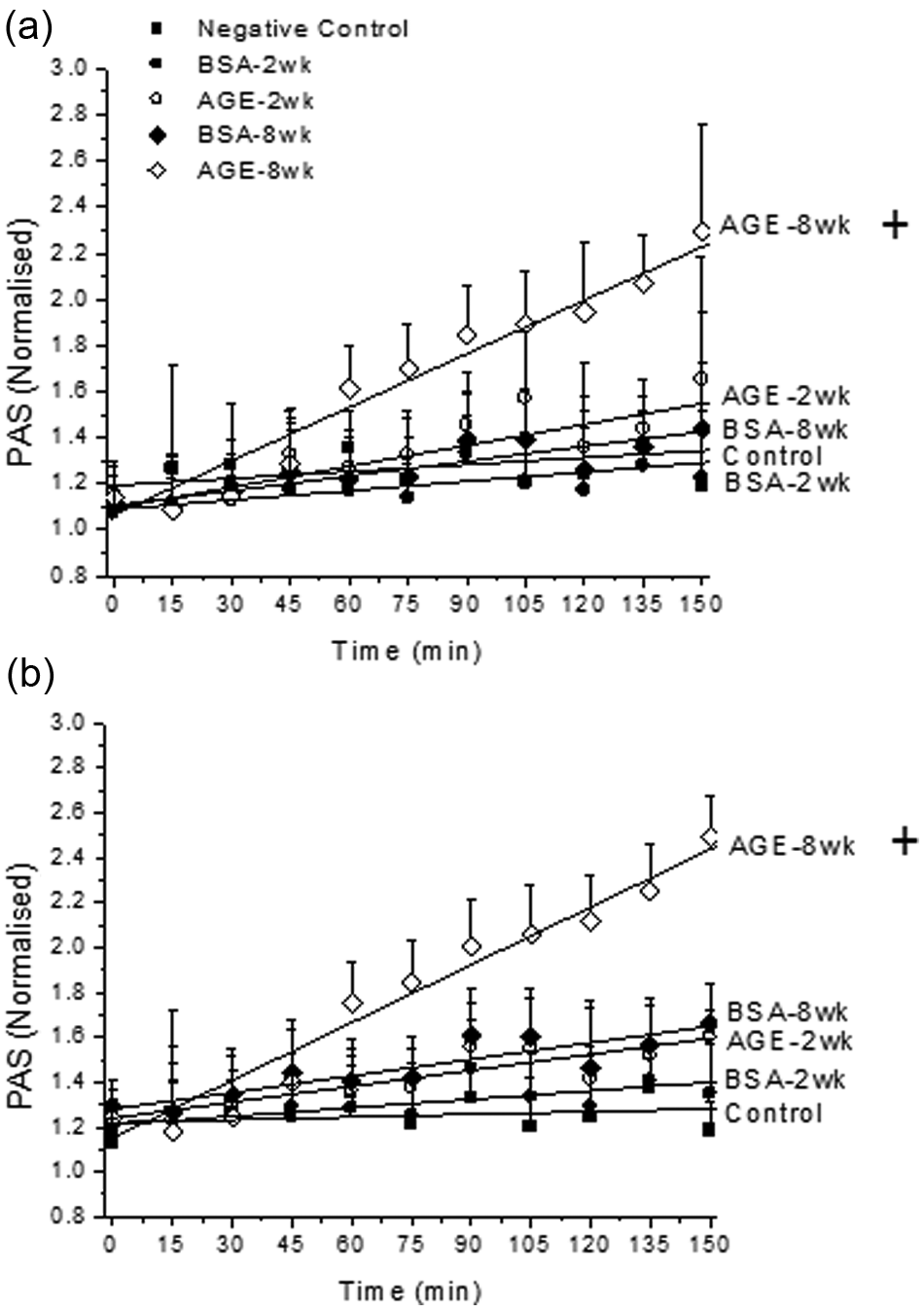
The platelet activation (PAS) rate in the presence of glycated (AGE) or non-glycated (BSA) albumin and mainstream (a) or sidestream (b) tobacco smoke extracts. Platelets were kept under static conditions for these experiments. The data shown represents the mean + standard error of the mean for six independent experiments. + Differs from paired BSA (by glycation time), BSA-2 week, AGE-2 week and negative control (p < 0.05, two-way ANOVA, paired by time).
Confirmation of the GPIIb/IIIa role in enhanced aggregation to tobacco smoke and advanced glycation end products
Timed platelet samples were removed and analysed for the surface expression of CD41 and CD62P to provide possible mechanisms for altered aggregation and activation responses. For aggregation studies, in response to the combined incubation of tobacco smoke extracts and glycated albumin, the expression of CD41 was significantly enhanced for all samples that were incubated with irreversibly glycated albumin and mainstream or sidestream tobacco smoke for 30 minutes or more (Figure 4(a), shown for sidestream extracts only for CD41, ranged from ~20–40% for all samples). For aggregation samples that exchanged nicotine for tobacco smoke extracts, the expression of CD41 was not different than the control conditions. Similarly, the enhancement in platelet activation, in response to the combined exposure to irreversibly glycated albumin and tobacco smoke extracts, was at least partially mediated by an enhanced fusion of α-granules to the platelet membrane, as quantified by CD62P expression. The enhancement varied from ~40% to 70% and increased nearly linearly over the 2.5 hour time course.
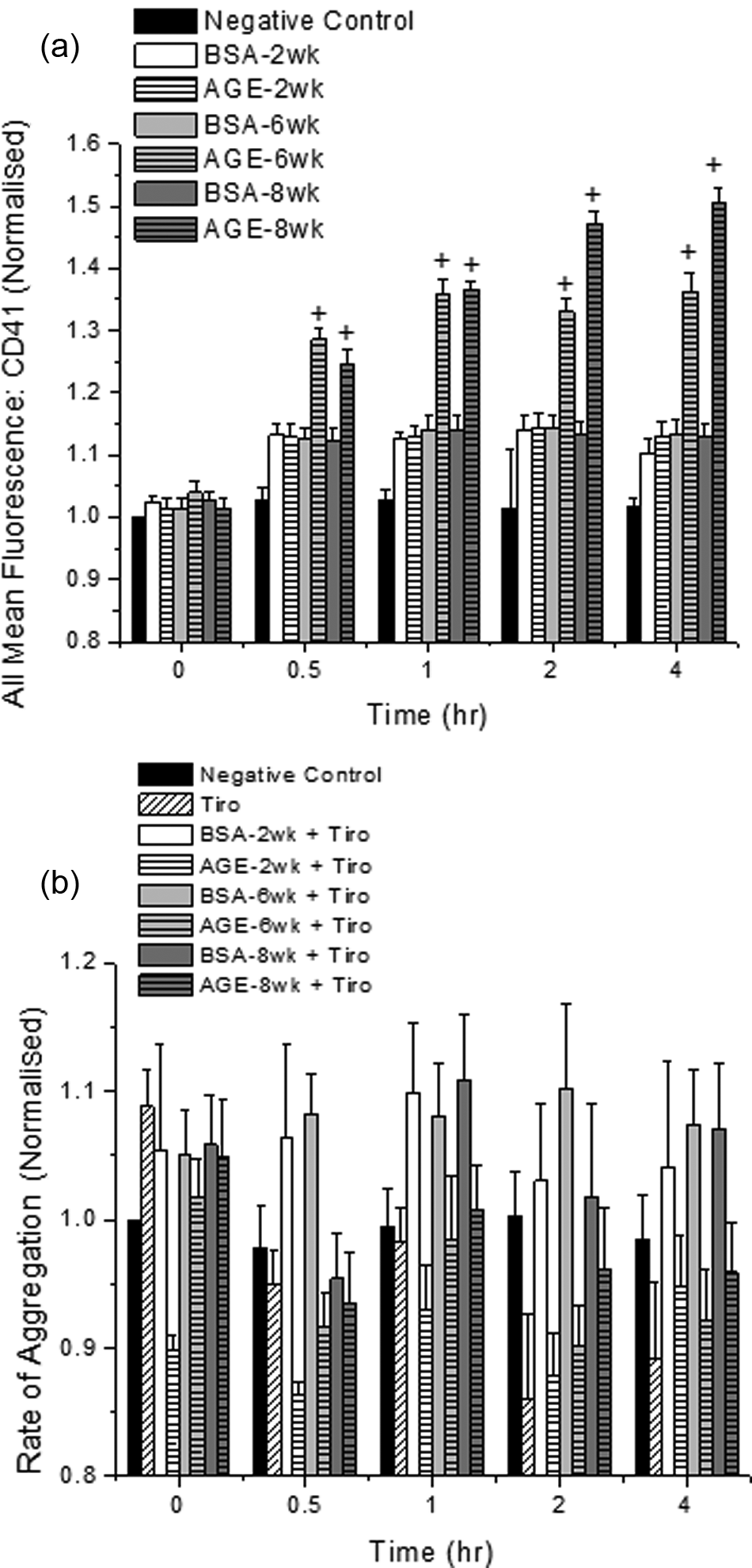
CD41 (GPIIb) expression (a) after incubation with sidestream tobacco smoke extracts and glycated (AGE) or non-glycated (BSA) albumin and the platelet aggregation response in the presence of 50ng/mL tirofiban (Tiro) (b). Platelets were kept under static conditions for these experiments. The data shown represents the mean + standard error of the mean for six independent experiments. + Differs from paired BSA (by glycation time), BSA-2 week, AGE-2 week and negative control (p < 0.05, two-way ANOVA, paired by time).
To confirm the role of GPIIb in the enhanced aggregation observed in response to tobacco smoke and advanced glycation end product incubation, platelets were incubated as described above with 50 ng/mL tirofiban. Our data shows that after blocking the effects of GPIIb, the aggregation response induced by tobacco smoke and advanced glycation end products is abolished and all aggregation returns back to control conditions (Figure 4(b), shown for sidestream extracts only). With the addition of mainstream tobacco smoke, the aggregation response was similarly reduced back to control conditions.
Discussion
Platelet aggregation
We hypothesised that combining cardiovascular risk factors would be more detrimental to platelet aggregation as compared with the individual risk factors or control conditions. Furthermore, we thought that the exposure to sidestream extracts would enhance platelet aggregation to a greater extent than mainstream extracts because sidestream smoke is the more potent (less diluted) form of tobacco smoke. Previous work from our group has showed that the individual incubation of platelets with glycated albumin cause an approximate 10–20% increase in the rate of platelet aggregation. 5 We have also looked into the individual incubation of platelets with sidestream smoke and have seen an approximate 10% increase in aggregation rate. 13 This data is in agreement with others that show sidestream tobacco smoke can enhance platelet aggregation responses.21,22 With regard to mainstream tobacco smoke effects on platelet aggregation (Figure 1(a)), previous reports show that there are minimal changes or possibly even inhibition of the aggregation response after exposure to tobacco smoke.23,24 Our data agrees with this, but it is important to point out that more physiologically relevant models (animal or clinical) have shown an increase in platelet aggregation after exposure to mainstream tobacco smoke, which is thought to be induced by changes to other cells that play a role during thrombosis, such as endothelial cells. 25 Here we show that by combining cardiovascular risk factors (Figure 1), the rate of platelet aggregation accelerates over the individual risk factor stimulation. We are not aware of other reports that investigated the platelet response to advanced glycation end products and tobacco smoke extracts, but others have linked accelerated cardiovascular disease progression, under these conditions, to endothelial cell changes. 12 Our flow cytometry data suggest that a possible mechanism for enhanced platelet aggregation is through the expression and activation of GPIIb (Figure 4(a)). This was confirmed through the addition of a low dose of tirofiban, a GPIIb inhibitor, which marginally inhibits the platelet aggregation response to TRAP6, but completely abolishes the aggregation response to tobacco smoke and advanced glycation end products (Figure 4(b)). Our results agree with others that have shown that in vivo, eptifibatide, another GPIIb blocker, can inhibit the deleterious cardiovascular events associated with cigarette smoking. 26 Therefore, it is likely that GPIIb is playing a salient role in cardiovascular disease progression through an enhanced expression stimulated by cardiovascular risk factors.
Role of nicotine
We also aimed to evaluate the role of nicotine on these processes, since: (a) nicotine is the major psychoactive component of tobacco smoke, (b) some smoking cessation programs rely on reducing the nicotine intake while maintaining the other cigarette components (e.g. low- to zero-nicotine cigarette programs), and (c) nicotine has an inhibitory effect in the hemostatic system. Our data has shown that with the addition of nicotine to either diabetic cardiovascular conditions or the combined diabetic and sidestream exposure conditions, platelet aggregation can be inhibited and that this is related to GPIIb expression (Figure 2 and flow cytometry). Although the exact mechanism by which nicotine acts on platelets is unknown, reports have shown that nicotine has an inhibitory role on platelet aggregation 27 and activation3,4 and that this may be mediated by beta-catecholaminergic stimulation of platelets. We have also shown that nicotine removal from cigarettes is very potent for platelet thrombogenicity. 3 If nicotine is shown to be inhibitory to other cardiovascular cells during disease progression, nicotine may be a potent molecule to deter cardiovascular disease progression.
Platelet activation
The remaining goal of this study was to identify the combined actions of advanced glycation end products and tobacco smoke extracts on platelet activation. We have previously shown that the individual incubation of platelets with either of these risk factors enhances platelet activation,4,5 by approximately 50%. In the present study, we saw that the combined incubation increased the platelet activation rate by over 100% and that this was partially mediated by increased α-granule fusion (P-selectin expression, Figure 3 and flow cytometry). Interestingly, under the combined incubation, the differences between mainstream extracts and sidestream extracts were abolished, which is suggestive of: (a) the potency of the combined risk factor stimulation, or (b) advanced glycation end products and some component(s) of the tobacco smoke extract compete and act through similar pathways. We are unaware of any work that has tried to tease these possible pathways out.
Summary and limitations
Here we assessed the platelet aggregation rate and the platelet activation rate for platelets subjected to diabetic cardiovascular conditions and tobacco smoke (mainstream or sidestream) or pure nicotine. The combination of cardiovascular risks (especially for sidestream extracts) significantly enhanced the aggregation and activation of platelets. This was partially mediated by an enhanced expression of CD41 and CD62P, respectively. The addition of pure nicotine reduced this potential. This has important implications for the diabetic population. It is already known that diabetes precedes many cardiovascular diseases, but with the exposure to tobacco smoke, whether intentional or not, this risk may be further enhanced, accelerating the progression of cardiovascular diseases. Furthermore, for those diabetic smokers that are currently on cessation plans, the beneficial role that nicotine plays in the cardiovascular system may be eradicated; particularly for those smokers who choose low- to zero-nicotine cigarettes. Our data would suggest that the cardiovascular risk potential for these cases would be even greater than the combined diabetic person exposed to tobacco smoke.
Although we have identified that the combination of tobacco smoke exposure and a diabetic vasculature can promote platelet activation and aggregation responses, which may lead to an enhanced cardiovascular disease progression, there are a number of limitations to our study that should be kept in mind. Our first major limitation is that washed platelets and platelet-rich plasma is not physiological. Furthermore, in vivo there are a number of other regulators that act on platelets, such as endothelial cells. Additionally, we use discrete populations of glycated albumin and fixed concentrations of tobacco smoke, whereas it is unlikely that a diabetic patient will only have reversibly glycated or irreversibly glycated albumin in their blood stream. Also, it is very difficult to mimic the lung extraction process and estimate the quantity of mainstream or sidestream tobacco smoke that is actually inhaled. However, even with those limitations, our data suggests a mechanism for enhanced platelet aggregation and platelet activity in response to combined cardiovascular risk factors.
Footnotes
Funding
This work was supported in part by the Oklahoma Center forthe Advancement of Science and Technology (grant no. HR09-158).
Conflict of interest
The authors have no competing interests to declare.