
Abstract
Diabetes is associated with increased risk of cardiovascular disease. Advanced glycation end-products (AGEs) are considered to be a major pathogenic factor for diabetic vascular complications. The levels of AGEs are increased in diabetic patients. We have studied the presence of the major AGE methylglyoxal (MGO)-derived hydroimidazolone in human aorta and carotid arteries, using immunohistochemistry (IHC), western blotting and mass spectrometry. By IHC, MGO-derived modifications were detected mainly associated with cells in intimal thickenings and cells in microvessels in adventitia. In type V lesions MGO-derived AGE was also present, extracellular in the necrotic core and in cells at the border of the core. The highest degree of modification was probably associated with cell nuclei. By western blotting and mass spectrometry fibrin(ogen), the cytoskeleton-associated protein moesin and the nuclear proteins lamin A and C were identified as putative main targets for MGO-derived modification. LC-MS/MS studies of fibrin(ogen) modified in vitro with low concentrations of MGO identified the sites that were most prone to modification. These results indicate that AGE modifications occur preferentially on specific proteins. The modification of these proteins may play a role in vascular dysfunction and development of atherosclerosis in diabetes.
Introduction
Cardiovascular disease is the major cause of mortality in diabetes. 1 It is well documented that diabetes contributes to accelerated development of atherosclerosis, 2 but the mechanisms behind this effect are not known in detail. There is, however, increasing evidence that advanced glycation end-products (AGEs) are important for this pathogenesis. 3 We have previously shown that measurements of ‘total’ serum AGEs predict cardiovascular mortality in women with type 2 diabetes. 4 AGEs are non-enzymatic modifications of proteins and lipids that result from complex reactions involving a combination of hyperglycaemia, hyperlipidaemia and oxidative stress. 5
The dicarbonyl compound methylglyoxal (MGO) is probably the most reactive intracellular AGE precursor. It is partly formed by dephosphorylation of triose phosphate intermediates of glycolysis, and it is catabolised to D-lactate by the glyoxalase pathway. Hyperglycaemia is shown to cause increased levels of MGO in endothelial cells. 6 The concentration of MGO in blood is 1 µM or less, while individuals with diabetes have 3–6 times this amount.7,8 There are, however, some variations in reported values, depending on methodology. MGO reacts with arginine residues on proteins to form 5-hydro-methylimidazolone (MG-H1) and, to a lesser degree, argpyrimidine. MG-H1 is one of the AGEs present at highest concentrations in human tissues and blood. 9 Recently it was shown that hyperglycaemia induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries, and that the impairment was mediated by increased intracellular MGO levels, 10 indicating that MGO may be a causative agent for endothelial dysfunction. A study of MGO-derived modifications of mitochondrial proteins in diabetic rats showed that only a subset of proteins was modified, and that these modifications were associated with excess superoxide formation. 11
MGO modifications occur extensively in the cell nucleus, and modifications of the transcriptional cofactors mSin312 and p30013 have been shown to be involved in dysfunction of endothelial cells and fibroblasts. MGO-derived modification of mSin3,12 caused by high glucose, was shown to be linked to increased transcription of angiopoietin-2 and increased proinflammatory effects in endothelial cells. MGO modification of p300 decreased the association between this protein and hypoxia-inducible factor-1α and impaired hypoxia-stimulated VEGF expression. It has been suggested 13 that this MGO-derived modification could partly explain the failure to form adequate compensatory microvasculature in response to ischaemia associated with diabetes.
Vascular disease in diabetes is associated with disruption of interactions between the extracellular matrix and endothelial cells, detachment of cells and premature cell death. 14 Integrins of endothelial cells and arginine–glycine–aspartate (RGD) and glycine–phenylalanine–hydroxyproline–glycine–glutamate–arginine (GFOGER) amino acid sequences of matrix proteins are important for these interactions. It has been shown that modification of the matrix protein collagen IV by MGO formed hydroimidazolones frequently at RGD and GFOGER sequences of this protein.15,16 The modifications caused cell detachment, apoptosis and impaired angiogenesis. It has also been shown that MGO modifications of the extracellular matrix impair the function of retinal capillary endothelium and its reparative potential in response to diabetes-related insults. 17 MGO-derived arginine modification of an RGD sequence in fibronectin was probably important for this impairment.
If specific proteins of the arterial wall are particularly prone to MGO modification, identification of these proteins may help to clarify the pathogenesis of AGE-dependent arterial lesions. The purpose of this paper is therefore to identify proteins in the arterial wall preferentially modified by AGEs (MGO modified).
Materials and methods
Autopsies were taken from 17 non-diabetic individuals (range 61–88 years, nine females and eight males), five diabetics (range 64–83 years, one female and four male) and a healthy child who suffered a sudden death (control). Four subjects died of cardiovascular disease, six subjects of cancer, three of pneumonia, while 12 subjects died of other reasons. Informed consent was obtained according to proven protocols established by law and in accordance with the Helsinki Declaration. The Regional Committee of Ethics in medical research approved the study.
The material consisted of predefined segments from the common carotid artery and the aorta. Consecutive slices of 1.5×0.5 cm2 were cut from small non-complicated atherosclerotic lesions and non-lesion areas and partly frozen to −76°C and partly fixed in 4% buffered formaldehyde. After formalin fixation, paraffin-embedded sections were stained with haematoxylin and eosin. Microscopically, sections with intimal thickening, containing foam cells, lipid-laden smooth muscle cells (SMCs) and extracellular lipid droplets (type III), 18 and atherosclerotic lesions with a fibrous cap and a necrotic core (type V) 19 were chosen for further examinations.
Immunohistochemical staining methods
Sections 4 µm thick from paraffin-embedded blocks were deparaffinised, rehydrated and immersed in citrate buffer (pH 6.0) in a Lab Vision PT module for 30 min for antigen retrieval. A hydrogen peroxide block for 10 min was used to reduce endogenous peroxidase activity. Ultra V block (Thermo Scientific, Fremont, CA, USA) for 5 min was used to reduce non-specific background. After blocking, the sections were incubated with a monoclonal mouse antibody against MGO imidazole AGE (1H7G5) 20 (1:200) for 15 min at room temperature followed by incubation with UltraVision ONE HRP polymer (Thermo Scientific) for 30 min at room temperature. To develop colour, a mixture of DAB Plus Chromogen and DAB Plus Substrate (Thermo Scientific) was added. Sections were counterstained with haematoxylin to locate cell nuclei. Normal IgG (Sigma, MO, USA) was used as a negative control. Immunostaining of macrophages and actin was performed with an Autostainer (HX System Benchmark, Ventana Medical Systems, Tuscon, AZ, USA). Mouse monoclonal antibodies against human macrophages (Macrophage, HAM-56) and smooth muscle isoforms of actin (Anti-Actin, Muscle, HUC 1-1) obtained from Ventana Medical Systems were used.
Extraction of proteins from arterial tissue, SDS-PAGE and two-dimensional electrophoresis
Tissues for protein studies were minced and extracted with 5 M urea in 50 mM Tris, pH 7.2, containing 10 mM EDTA. 21 The protein concentration was determined by the Bradford method. 22 Proteins were separated by SDS-PAGE, using NuPAGE 4–12% Bis–Tris gels (Invitrogen, Carlsbad, CA, USA). In two-dimensional electrophoresis the proteins were first separated by isoelectric focusing (IEF) using Immobilone DryStrip pH 3–10, 7 cm (GE Healthcare, Buckinghamshire, UK) which was rehydrated in 7 M urea, 2 M thiourea, 4% CHAPS, 0.2% Pharmalytes and 1.5 mg/ml DTT. IEF was run with a Bio-Rad Protean IEF Cell (Bio-Rad, Hercules, CA, USA). After focusing the strips were equilibrated in LDS sample buffer (Invitrogen) containing reducing and alkylation solutions and the proteins were applied to SDS-PAGE. Proteins were visualised by staining with Coomassie Brilliant Blue G-250 (Sigma). Magic Mark protein standard (Invitrogen) was used to estimate molecular weights.
Western blotting
After SDS-PAGE, proteins were transferred to an Immobilon-P membrane at 35 V for 60 min using NuPAGE transfer buffer (Invitrogen). The membranes were incubated for 1 h in Protein-Free T20 (TBS) Blocking Buffer (Pierce, Rockford, IL, USA) and then incubated overnight at 4°C with a primary antibody. The following antibodies were used: a monoclonal mouse anti-MGO-derived imidazole AGE (1H7G5) 1:3,000, 20 a polyclonal anti-lamin A/C: sc-20681 (Santa Cruz, CA, USA), and a monoclonal mouse anti-albumin: sc-51515 (Santa Cruz). Peroxidase-conjugated goat anti-mouse and anti-rabbit IgG (1:10,000, Jackson ImmunoResearch laboratories, Inc., West Grove, PA, USA) were used as secondary antibodies. An enhanced chemiluminescence system (GE Healthcare) was used together with a CCD-camera (Chemigenius Bio Imaging system, Syngene, Cambridge, UK) to measure peroxidase activity.
Incubation of fibrin(ogen) with MGO in vitro
Fibrin(ogen) from human plasma, essentially plasmin(ogen) free, (Sigma) at 1.3 mg/ml was incubated with MGO (1–500 µM) at 37°C for 24 h and applied for either SDS-PAGE and western blotting or LC-MS/MS studies.
Protein identification by LC-MS/MS
The Coomassie Brilliant Blue G-250 (Sigma) gel spots were excised for in-gel digestion with 0.1 µg trypsin (Promega, Madison, WI, USA) in 20 µl 25 mM ammonium bicarbonate, pH 7.8 at 37°C for 16 h. For each band the tryptic peptides were purified with µ-C18 ZipTips (Millipore, Billerica, MA, USA). Dried peptides were dissolved in 10 µl 1% formic acid in water, and a volume of 5 µl was injected onto an LC/MS system consisting of a Dionex Ultimate 3000 nano-LC system (Sunnyvale CA, USA) connected to a linear quadrupole ion trap–Orbitrap (LTQ Orbitrap) mass spectrometer (ThermoElectron, Bremen, Germany) equipped with a nanoelectrospray ion source. For liquid chromatography separation we used an Acclaim PepMap 100 column (C18, 3 µm, 100 Å) (Dionex, Sunnyvale CA, USA) capillary of 12 cm bed length. The analysis was performed at the Proteomics Core facility of the University of Oslo at the Biotechnology Centre of Oslo.
The mass spectrometer was operated in the data-dependent mode to automatically switch between Orbitrap-MS and LTQ-MS/MS acquisition. The method used allowed sequential isolation of the most intense ions, up to six, depending on signal intensity, for fragmentation on the linear ion trap using collisionally induced dissociation at a target value of 100,000 charges.
For accurate mass measurements the lock mass option was enabled in MS mode. 23 Further details about instrument parameters and data analysis have been described elsewhere. 24
MALDI-TOF analysis
Protein in Coomassie-stained gel slices was reduced with DTT (GE Healthcare), alkylated with iodoacetamid (Sigma Aldrich) and cleaved by trypsin (V511A, Promega). Peptides were extracted with trifluoroacetic acid and acetonitrile and analysed by MALDI-TOF (Ultraflex ToF/ToF, BRUKER Daltonic GmbH) by the Proteomic Unit at University of Bergen, Norway. Mascot search in the database NCBlnr was performed to identify the peptides.
Data analysis for detection of modified amino acids
Proteins were identified by automated searching of all MS/MS spectra using the SEQUEST algorithm incorporated in Proteome Discoverer software (Thermo Scientific). Only the peptides resulting from the tryptic cleavages were searched and two trypsin missed cleavages were allowed. Carbamidomethylation of cysteine (57.02 Da) was selected as a fixed modification, while MGO modification at arginine (54.05 Da) and oxidation of methionine (15.99 Da) were selected as variable modifications for all searches. All searches were performed with a precursor mass tolerance of 10 ppm, and SEQUEST results were filtered using the following filters: Xcorr ≥ 1.9, 2.2 and 3.75 for singly, doubly and triply charged ions, respectively.
Results
Immunohistochemical staining
The MGO-derived imidazolone AGE staining of a common carotid artery of a child, with an adaptive intimal thickening, is shown in Figure 1. Slight positive staining of MGO-derived imidazolone (1A and 1B) was obtained in intima close to the lumen and was mainly intracellular. Only faint and diffuse staining was obtained with normal IgG, which was used as a negative control (1C).
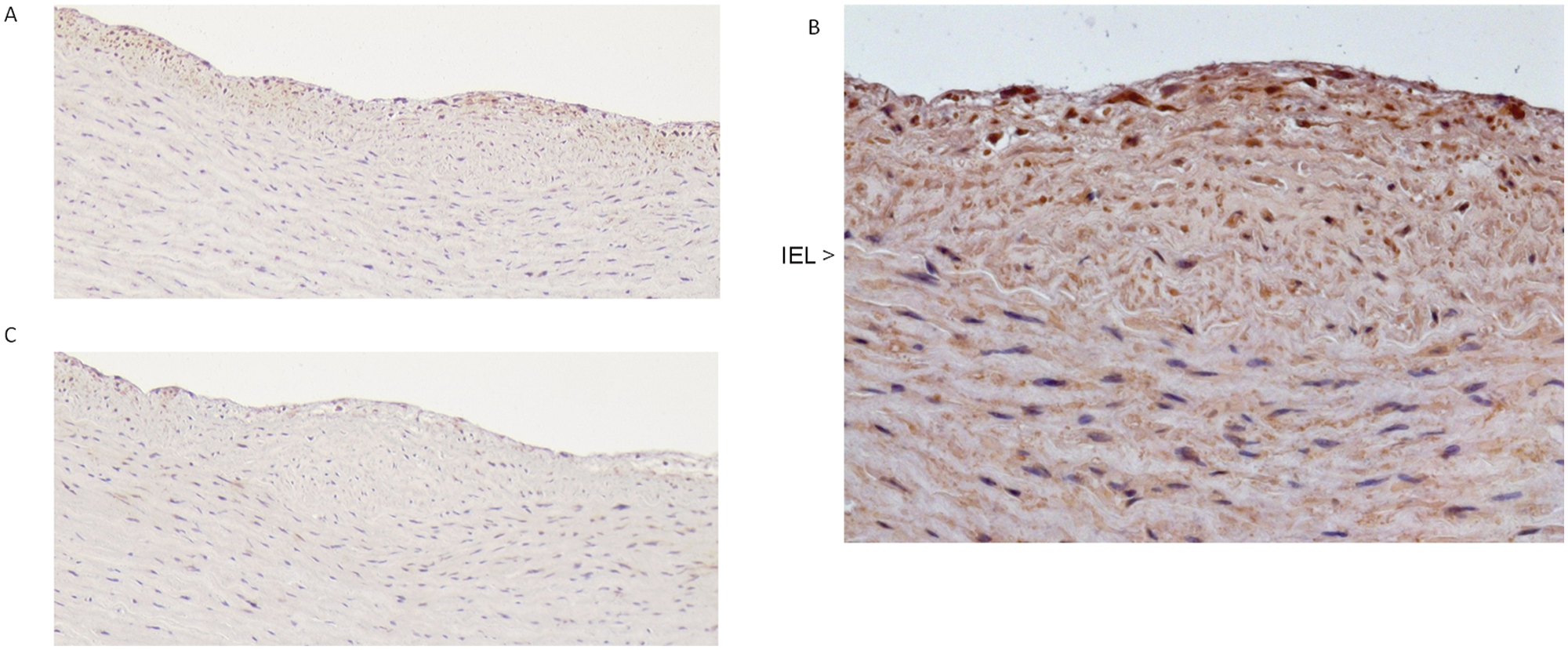
Representative immunostained section from an adaptive intimal thickening taken from the common carotid artery of a healthy child who died in an accident. Intima is without foam cells, contains spaced smooth muscle cells and is distinctly separated from media by internal elastic lamina (IEL). Positive staining of MGO modifications was obtained intracellular in intima close to lumen (A ×100, B ×400). Staining obtained with normal IgG was used as a negative control (C ×100).
MGO-derived imidazolone staining of a representative type III atherosclerotic lesion is shown in Figure 2A and 2B. Strong staining was associated with most of the cells in the intimal thickening and in cells near the lipid pools, while fainter staining occurred in media. The staining in intima overlapped with staining for SMC (Figure 2C), while the staining near the lipid pools overlapped with staining for macrophages (Figure 2D). MGO staining of an atherosclerotic type V lesion is shown in Figure 3. The section contained a fibrous cap and a necrotic core with cholesterol crystals (Figure 3A). Strong positive MGO-derived AGE staining was associated with cells in the fibrous cap and cells surrounding the necrotic core (Figure 3B–D). Several of the latter cells were foam cells (Figure 3D), and the staining was particularly strong in the nuclei of these cells. Positive extracellular MGO imidazolone staining occurred in the necrotic core (Figure 3C and 3D). Furthermore, positive staining with the MGO antibody was associated with nuclei of endothelial cells in microvessels in adventitia (Figure 3E). This staining in adventitia occurred in variable strength in all tissue that was examined.

Immunostaining for MGO-derived AGE, SMC and macrophages in aorta of an 80-year-old man. The section is from an atherosclerotic type III lesion as shown by small lipid pools and layers of smooth muscle cells. Positive MGO staining appears associated with cells both in intima and media (A ×100, B ×400). Staining of consecutive sections for smooth muscle actin (C ×100) and macrophages (D ×400) shows overlap with MGO-positive cells.
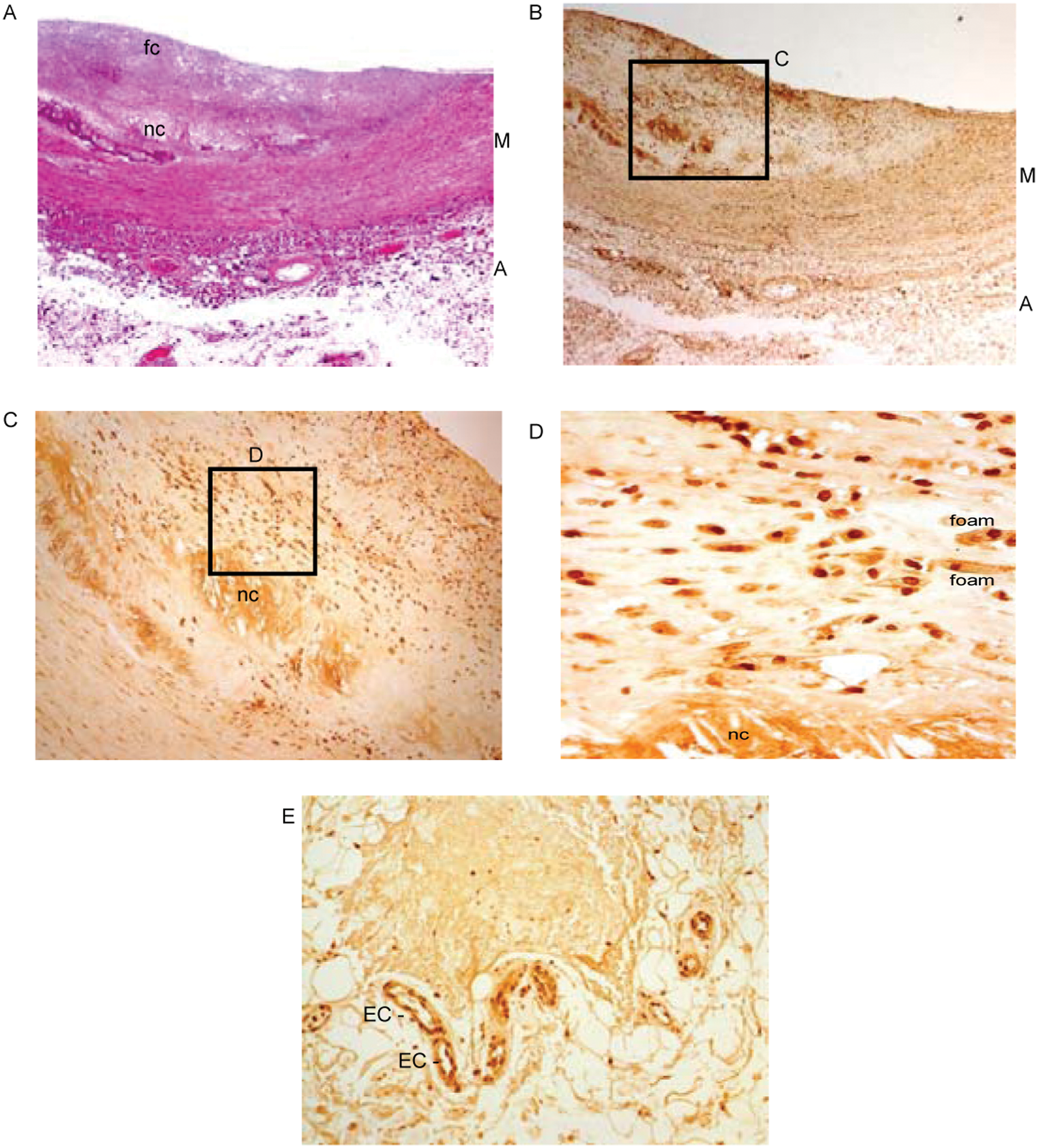
Immunostaining of an atherosclerotic type V lesion in a common carotid artery from a 76-year-old man. H-E stained section shows the presence of a fibrous cap (fc) and a necrotic core (nc) (A ×25). A consecutive section immunostained for MGO-derived AGE shows strong positive staining associated with cells in the fibrous cap and the upper border of the necrotic core (B ×25, C ×100, D ×400). Several of the latter cells were foam cells (foam). Positive staining is also seen extracellular in the necrotic core (D) and in endothelial cells (EC) of vasa vasorum in adventitia (E ×200).
Identification of MGO-derived imidazole-modified proteins by western blotting
Protein samples were fractionated by SDS-PAGE and 2-D electrophoresis followed by Coomassie staining or western blotting using an antibody against MGO AGE (Figure 4A–4D). Proteins with an apparently high ratio of western staining to Coomassie staining were cut out of a Coomassie-stained gel and analysed by mass spectrometry. Fibrin(ogen) α-chain (labelled 2–4) and lamin A (labelled 6) were identified by LC-MS/MS (with Mascot probability <0.05), while fibrin(ogen) β-chain (labelled 1) and moesin (labelled 5) were identified by MALDI-TOF (with Mascot probability <0.05). Western blotting with an antibody against lamin A/C showed that these proteins overlapped with several spots from western blotting with the MGO AGE antibody (Figure 4E). Fibrin(ogen) α-chain, moesin and lamin A gave very faint spots when stained with Coomassie (Figure 4D), indicating that the specific degree of modification might be high for these proteins. Analysis of samples from different parts of carotid artery and aorta from different persons showed great variation in the level of fibrin(ogen) (results not shown). The protein in the strongest Coomassie-stained band at 65 kDa was identified as albumin by a specific antibody. The 2-D gel electrophoresis results, when taken together, revealed that several strong bands seen in 1-D SDS-PAGE (Figure 4A) were due to a mixture of different proteins. A sample from a child gave much fainter signals from most of the identified proteins when analysed by western blotting with the MGO AGE antibody (Figure 4F).
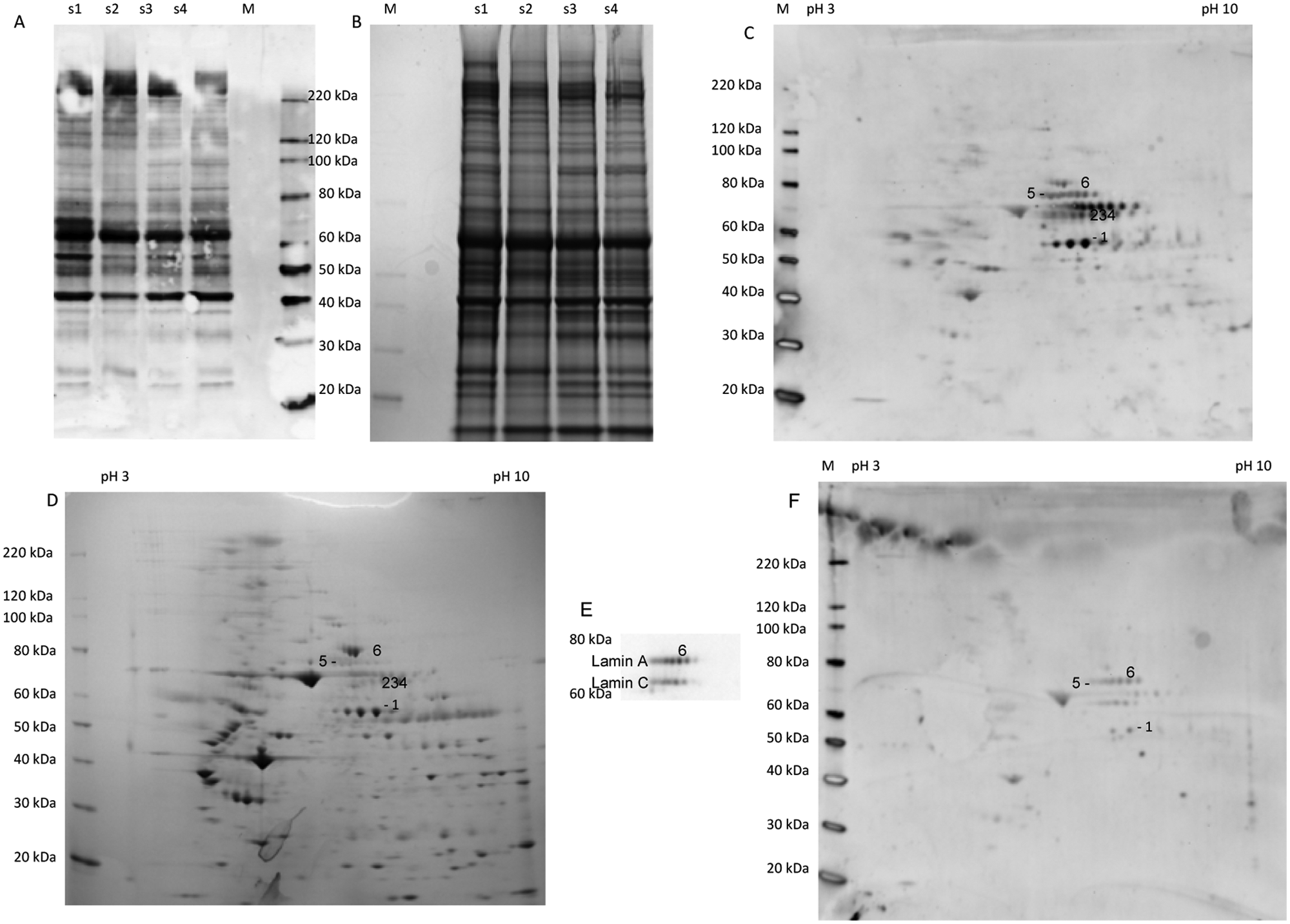
MGO-derived modifications of proteins extracted from different tissue samples. Samples s1 and s2 were from aorta, and s3 and s4 were from the common carotid artery. The samples (2 μg) were analysed by 1-D SDS-PAGE followed by western blotting with an antibody against MGO-derived AGE (A). Another gel with the same samples (30 µg) was stained with Coomassie blue (B). Sample s1 (15 μg) was also analysed by 2-D, with an isoelectric focusing gradient from pH 3–10, followed by western blotting with the same antibody (C). Identified proteins are labelled by specific numbers. The protein in spot 1 was identified as the β-chain of fibrin(ogen), the protein in spots 2–4 as the α-chain of fibrin(ogen), the protein in spot 5 as moesin and the protein in spot 6 as lamin A. Proteins labelled 2–4 are just above the numbers, while the protein labelled 6 is just below the number. Another 2-D gel with the same sample (45 µg) was stained with Coomassie blue (D). A specific antibody was used to identify different forms of lamin A and C (E). A sample (15 μg) from a young person with normal intima was also analysed by western blotting (F). Magic mark (M) was used to estimate molecular weights.
Modification of fibrin(ogen) by MGO in vitro
Fibrin(ogen) from human plasma was used for in vitro studies with MGO. LC-MS/MS showed that this product contained some fibronectin in addition to the α, β and γ-chains of fibrin(ogen). Plasma fibrin(ogen) was incubated with MGO concentrations ranging from 5 μM (slightly above that found in diabetics) to 500 μM for 24 h. The latter concentration has been used in similar experiments by others15,16 and yields modification of approximately one arginine per molecule of albumin. At the highest MGO concentrations hydroimidazolone was detected at many sites, especially in the α and β-chains (Figure 5), while it was detected only at a few sites in the same chains at the lowest concentrations. Altogether, 22 MGO-modified peptides were identified for α- and β-chain by their collision-induced dissociation MS/MS fragmentation patterns (Supplementary material Table S1). MS/MS fragment ion spectra of peptide MELErPGGNEITR derived from α chain is shown in Figure S1 (Supplementary material). Modification of the RGD site in the C-terminal part of the α-chain, which is involved in interactions with integrins of endothelial cells, was detected at and above 20 μM MGO. No modification of RGD sites of fibronectin was detected.

Fibrin(ogen) from human plasma was incubated with 5, 10, 20, 50, 100, 200 and 500 μM MGO, respectively, for 24 h. Sites in the α- and β-chains where hydroimidazolone was detected by LC-MS/MS are in bold and underlined. Most of the sites were detected when concentrations at and above 50 µM MGO were used. Only a few sites (indicated by small letters) were detected when 5 µM MGO was used.
Fibrin(ogen) from human plasma, modified by MGO, was also analysed by SDS-PAGE and western blotting using the antibody against MGO hydroimidazalones (Figure 6). In the western blot (Figure 6B) two different forms of the α-chain gave the strongest signals when no MGO was added (labelled 0). Fibrin(ogen) modified by 1 µM MGO (labelled 1), which is close to physiological concentrations, gave a slightly stronger signal than untreated fibrin(ogen), and the signal strength increased with increasing concentrations of MGO (Figure 6B), indicating that the antibody could be used for semi-quantification of MGO modifications of this protein. Proteins extracted from aorta (t, t1 and t2) were used as a comparison. A direct comparison of the degree of modifications in this in vivo sample and fibrin(ogen) modified in vitro is, however, difficult since other proteins in the former sample overlap with fibrin(ogen).
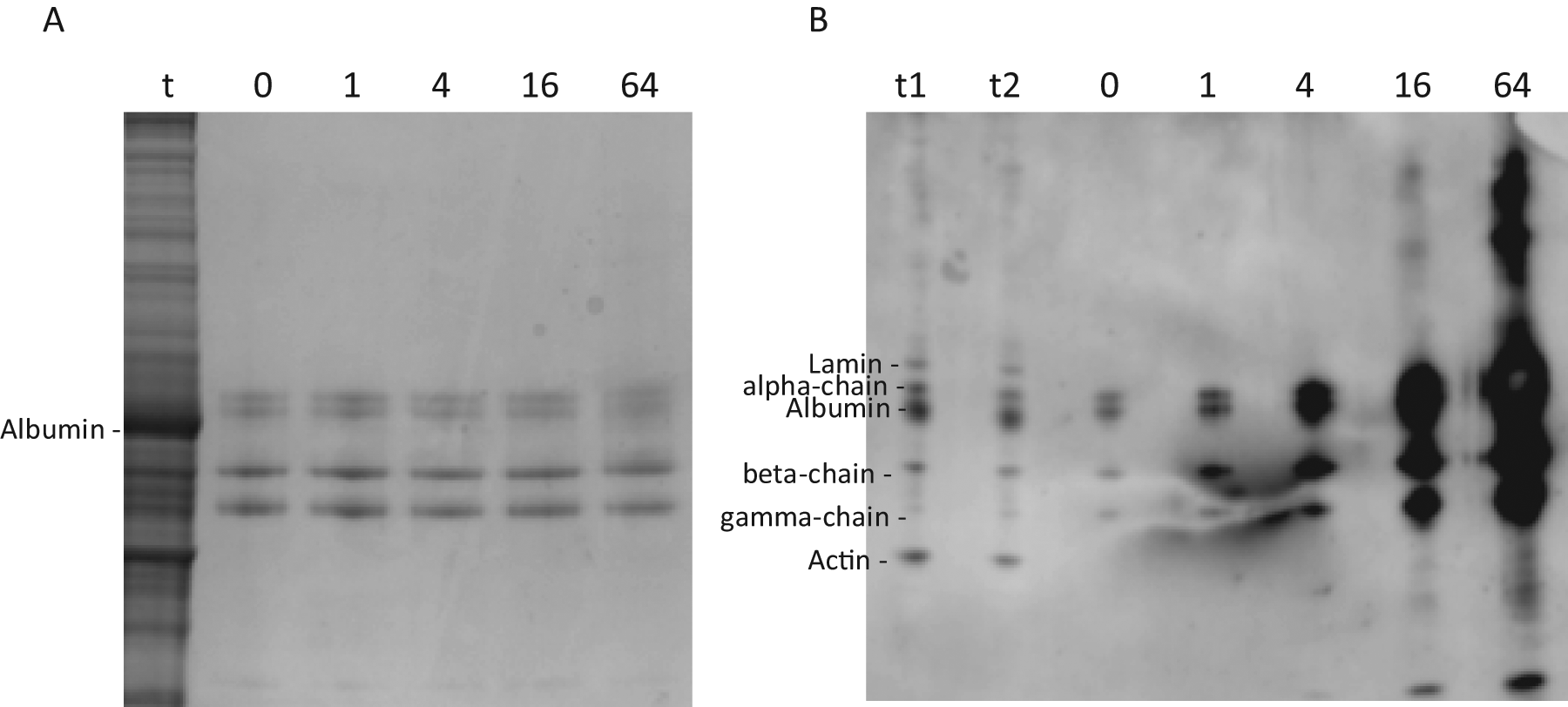
Fibrin(ogen) from human plasma was incubated with 1, 4, 16 and 64 µM MGO, respectively, for 24 h. After incubation fibrin(ogen) was analysed by SDS-PAGE (A) (1.7 µg) and western blotting (43 ng), using the antibody against MGO-derived hydroimidazolones (B). A tissue sample in two different concentrations (t1 and t2) and untreated fibrin(ogen) (0) were also included. The concentrations of MGO are indicated in µM. The upper fibrin(ogen) bands probably contain two different forms of the α-chain. The lower of these bands overlapped strongly with albumin.
Discussion
The aim of the present work was to localise MGO- derived hydroimidazolone, a major AGE, in arteries, and to identify putative main protein targets for this modification. Fibrin(ogen) was identified by western blotting and LC-MS/MS as a putative main target for MGO modification. Fibrin(ogen) α-chain was identified in three different spots that corresponded to spots in western blotting with the MGO AGE antibody, ensuring the identity of the modified protein. The α-chain seems to be extensively modified compared with other proteins, since it was scarcely visible by Coomassie staining in contrast to high MGO AGE immunoreactivity. Hydroimidazolone was identified on fibrin(ogen) when the protein was modified in vitro at MGO concentrations similar to those occurring in vivo.
It is well established that fibrin(ogen) can induce vascular dysfunction during cardiovascular disease. 25 Early atherosclerotic plaques contain fibrin(ogen), present as long threads surrounding cells. 26 High levels of fibrin have been found in necrotic cores of coronary arteries proportional to intraplaque vasa vasorum, and it was proposed that it may play a role in the development of atheroma from pre- atheroma. 27 Fibrin(ogen) has also been detected within a subset of intimal microvessels and was associated with inflammation and cellular infiltration. 28 This is particularly interesting as diabetic plaques have increased neovascularisation. 29 Fibrin(ogen) induces attachment, spreading and migration of endothelial cells 30 dependent on an RGD site in the C-terminal part of the α-chain which interacts with specific integrin cell receptors. 25 Similar RGD sites in type IV collagen15,16,31 and fibronectin 17 are shown to be particularly susceptible for modification by MGO, and the interaction between immobilised collagen IV and fibronectin and endothelial cells was impaired when these proteins were modified in vitro by 0.1–2 mM MGO. In the present work hydroimidazolone at the RGD site of the C-terminal end of the fibrin(ogen) α-chain was detected when the protein was incubated with 20 μM MGO. It could therefore be suggested that MGO modification of the α-chain of fibrin(ogen) might impair the interaction between this protein and endothelial cells in vivo. MGO modification may also influence fibrinolysis, since a cleavage site for plasmin between R491 and H492 of the α-chain (Figure 5) involves an arginine that was modified in vitro by 5 μM MGO. Hydroimidazolone was also detected in vitro at R23 of the fibrin(ogen) β-chain. R23 is in a part of the molecule that interacts with VE-cadherin. This interaction is probably important for endothelial cell spreading, capillary tube formation and angiogenesis. 32 It has also been suggested that the interactions between VE-cadherin and fibrin(ogen) are involved in the regulation of the transmigration of leukocytes through endothelial surfaces. 33
Transient interactions between endothelial cells and a provisional matrix composed mainly of fibrin, fibronectin and vitronectin occur during neoangiogenesis, and integrin αvβ3 is expressed at this stage of the process. 34 It is therefore likely that MGO modification of RGD sites of fibrin(ogen) (and fibronectin) will impair neoangiogenesis in diabetes. Interestingly, it has been shown that MGO modification of fibronectin impairs the function of the retinal capillary endothelial cells and their reparative potential. 17
Moesin and lamin A, two other apparent main MGO targets, appeared close to each other after 2-D electrophoresis. The presence of MGO-modified moesin was further verified after fractionation based on affinity to heparin (not shown). The presence of MGO-modified lamin A was additionally verified by an antibody against this protein (Figure 4E). The cytoskeleton-associated protein moesin serves as a cross-linker between the plasma membrane and actin filaments. This protein is important for the recruitment of leukocytes from the blood to the vessel wall by coordinating the functions of the cytokine-induced adhesion molecules E-selectin, ICAM-1 and VCAM-1. 35 An MGO-derived modification of moesin may therefore contribute to endothelial dysfunction and thus be involved in atherogenesis.
AGE modifications occur extensively in the cell nucleus, and MGO modifications of the transcriptional cofactors mSin312 and p30013 have been shown to be involved in dysfunction of endothelial cells and fibroblasts. Lamin A and C, two proteins of the inner part of the nuclear envelope, were shown to be putative main targets for MGO modifications. Lamin A and C provide a structural framework for the nucleus, but are also involved in chromatin organisation, DNA replication, transcription, and DNA repair. 36 Post-transcriptional modifications are important for the regulation of their function. Mutations in the LMNA gene, encoding lamins A and C, are responsible for Hutchinson–Gilford progeria which is characterised by severe premature atherosclerosis. 37 MGO modification of lamin A/C may therefore be of importance for the development of atherosclerosis.
Immunohistochemistry showed that MGO-derived AGEs were associated with endothelial cells in microvessels in the adventitia of aorta and carotid arteries. This is partly in accordance with a recent study showing the presence of MGO-derived hydroimidazolone in the adventitia and endothelium of mesentric arteries in rats. 10 Strong staining was also associated with SMCs in the intimal thickening of type III lesions and the fibrous cap of type V lesions. Dysfunction of SMCs in the intima caused by MGO modification will be pathologically important since these cells synthesise most of the structural proteins and enzymes in this part of the vessel wall. Strong staining for MGO modification was associated with inflammatory cells close to the necrotic core in type V lesions. This area is often associated with ischaemia, high levels of reactive oxygen species and frequent cell death. MGO modification in these cells may cause cellular dysfunction, and enhanced necrosis and apoptosis. Extracellular MGO-derived AGE was present in necrotic cores and may be due to modification of proteins that had leaked from microvessels. It is likely that much of the extracellular MGO-derived AGE is due to MGO-modified fibrin(ogen). Fibrin(ogen) was identified by western blotting and LC-MS/MS as a possible main target for MGO modification and has been detected at high levels in necrotic tissue. 27 Particularly strong staining was associated with cell nuclei. Recent studies have shown concentrated MGO-derived hydroimidazolone around the nuclei of endothelial cells grown in the presence of MGO, 10 and MGO-derived argpyrimidine was present exclusively in the nucleus of neural cells. 38 MGO modification of lamin A/C of the nuclear envelope may contribute to the nuclear immunohistochemical staining pattern in the present work, since western blotting and mass spectrometry showed that these proteins may be main targets for MGO modification.
There are limitations to this study. A quantitative estimation of the degree of MGO modification of fibrin(ogen) in different samples was not given. Furthermore, moesin and lamin A overlapped in 1-D gels. We could therefore not present a quantitative comparison of MGO modification of fibrin(ogen), lamin A and C and moesin from diabetics and non-diabetics. MGO-derived hydroimidazolone is, however, generally increased in diabetics. 39 It is therefore highly probable that differences occur in vivo in the level of MGO modification of fibrin(ogen), lamin and moesin between diabetics and non-diabetics. Urea and EDTA were used to extract proteins for western blotting. 21 Collagen type I and III and elastin are not solubilised under these conditions, and we did not investigate putative MGO modification of these proteins.
Conclusions
Putative main targets for MGO-derived AGE modification in aorta and carotid arteries of humans were identified for the first time. MGO-derived modifications of fibrin(ogen) may have consequences for fibrinolysis and interactions between fibrin(ogen) and endothelial cells. Modifications of fibrin(ogen), moesin and lamins may be part of the mechanism that leads to enhanced vascular dysfunction and atherosclerosis in diabetics.
Footnotes
Acknowledgements
We thank Dr Michael Brownlee (Albert Einstein College of Medicine, Bronx, NY, USA) for the gift of the monoclonal antibody 1H7G5.
This study has been funded by Medical Faculty (Thematic area Diabetes), University of Oslo, Regional Health Authority (Helse Sør-Øst), Diabetes Research Fund, Aker and Ullevål University Hospitals and Hormone laboratory, Oslo University Hospital, Aker.
The authors declare that they have no conflict of interest.