
Abstract
Background
Sepsis-associated acute kidney injury (SA-AKI) is marked by systemic inflammation and organ dysfunction. Ubiquitin-specific protease 18 (USP18) is a regulator in immune responses and apoptosis.
Objectives
To explore USP18's role in inflammatory pathways and apoptosis in SA-AKI, particularly its interactions with the PI3K-AKT-NF-κB pathway.
Design
A combination of animal experiments and cellular models was employed to investigate the expression and regulatory functions of USP18 in both in vivo and in vitro settings.
Methods
We established SA-AKI models in mice and HK-2 cells using LPS. Gene expression was analyzed via RNA-seq, and Cr, BUN, and inflammatory factors were measured using biochemical assays and ELISA. Kidney pathology was assessed with HE staining, mRNA levels with qRT-PCR, protein expression with Western blots, and cell apoptosis with flow cytometry.
Results
In SA-AKI mice model and HK-2 cell lines, upregulated USP18 was linked to increased activity in the PI3K-AKT-NF-κB pathway and heightened inflammation. Conversely, USP18 downregulation decreased early cell apoptosis and raised levels of inflammatory proteins. Elevated USP18 levels were also found in SA-AKI patients’ blood and urine.
Conclusion
USP18 enhances in vitro and in vivo responses by modulating inflammation and apoptosis through the PI3K-AKT-NF-κB pathway, presenting a potential target for SA-AKI therapy.
Introduction
Acute kidney injury (AKI) is a common disease in the ICU, and sepsis associated acute kidney injury (SA-AKI) is a common type. Sepsis, the systemic inflammatory response to infection, involves a dysregulated immune reaction that can lead to tissue damage and organ dysfunction, including AKI.1,2 During sepsis, the body's immune system mounts an exaggerated response to the infection, releasing pro-inflammatory cytokines and activating various signaling pathways, including the PI3K-AKT-NF-κB pathway. This inflammatory cascade can disrupt normal cellular functions in the kidneys, leading to cell apoptosis. 3
In previous pathway studies, the classic PI3K-AKT-NF-κB pathway was shown to play a role in regulating immune and inflammatory responses.4–6 Numerous pharmacological studies have indicated that inhibiting PI3K/AKT can ameliorate kidney injury,7,8 thus indicating its promise as a potential clinical biomarker or therapeutic target. However, it remains unknown the implication of this cell signaling in the pathogenesis of SA-AKI.
Ubiquitin-specific protease 18 (USP18) is an important member of the ubiquitin-specific protease family and a protein with multiple functions.9,10 It is an isopeptidase that inhibits the interferon signaling pathway. 11 Several studies have highlighted the involvement of USP18 in LPS-induced inflammation, including sepsis.12–14 In recent years, studies have revealed that USP18 is associated inflammatory reactions and cell apoptosis by interference in several pivotal pathways, especially PI3K-AKT-NF-κB.15–18 However, its specific contributions through this cardinal pathway to SA-AKI remain incompletely understood.
Currently, clinical tests commonly used to assess kidney damage involve measuring blood creatinine (Cr) and blood urea nitrogen (BUN) levels. 19 Recently discovered biomarkers of AKI include NGAL, cystatin C, KIM-1, IL-18, IGFBP-7, and TIMP-2.20,21 However, due to the multifaceted nature of SA-AKI, these biomarkers have proven ineffective in reliably diagnosing SA-AKI patients and accurately assessing the severity and progression of the condition.
Given the pivotal roles of USP18 in inflammation, we hypothesized that USP18 could serve as a potential biomarker for SA-AKI. In this paper, our aim is to elucidate the intricate interplay between USP18, the PI3K-AKT-NF-κB pathway, and the pathogenesis of SA-AKI. We aim to provide insights into the mechanisms underlying renal injury in the context of sepsis, with a specific focus on the regulatory roles of USP18 and the PI3K-AKT-NF-κB pathway. By unraveling these molecular complexities, we endeavor to lay the groundwork for the development of novel diagnostic biomarkers and therapeutic strategies aimed at mitigating renal injury and improving clinical outcomes in SA-AKI patients.
Material and methods
Mouse feeding and modeling
22 C57BL/6 male mice (wild-type) were obtained from Weitong Lihua Experimental Animal Technology Co., Ltd. Beijing. The mice were divided into the control group (10 mice) and LPS model group (12 mice), adaptively housed for 5–7 days, during which their health and behavior were monitored closely. This observation period served to ensure the mice's suitability for the subsequent construction of a SA-AKI mouse model. To induce sepsis, the model group mice received an intraperitoneal injection of LPS at a dosage of 10 mg/kg.22,23 The control group received an intraperitoneal injection of saline solution. Following administration, the mice were kept under standard feeding conditions for an additional 24 h. Subsequently, the mice were then euthanized to verify the successful establishment of the SA-AKI model according to the serum levels of Cr, BUN and inflammatory factors. This study was approved by the Ethics Committee of the First Affiliated Hospital of Harbin Medical University (2022080).
Cell culture and modeling
Human renal tubular epithelial cells (HK-2) were obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HK-2 was cultured in DMEM-F12 complete medium (GUMD-R104) and maintained at 37 °C in humidified incubator which contains 5% CO2. The culture medium was refreshed every 48 h to ensure optimal cell growth. Upon reaching 70%–80% confluency, HK-2 cells were subjected to modeling by exposure to LPS at different concentrations (1 μg/ml, 5 μg/ml, and 10 μg/ml). After evaluating the models by the levels of Cr, BUN and inflammation factors, a final decision was made to utilize the 10 μg/ml concentration for subsequent experiments.
Detection of Cr, BUN, and inflammatory factors in mice
Upon euthanasia of the mice, blood samples were collected from the eyeballs, and plasma was subsequently obtained via centrifugation. Cr and BUN levels were determined using creatinine and urea nitrogen test kits (C013-2-1, Nanjing). Additionally, ELISA kits were utilized to measure the levels of IL-6 (YJ002293, Mlbio) IL-1β (YJ301814, Mlbio) and TNF-α enzyme-linked immunosorbent assay (JLC3924, Mlbio). Experimental protocols were followed accordingly.
Hematoxylin-Eosin (HE) staining of mice kidneys
Fixed kidney tissues were embedded, dried, and sectioned before being subjected to HE staining. Following HE staining, morphological changes and the presence of inflammatory exudates in the kidneys of both the control and model groups were observed under microscopes at magnifications of 40×, 100×, 200×, and 400×.
RNA extraction and RNA-Sequencing
Kidney samples from three mice in both the control and model groups were subjected to RNA sequencing by OE Biotech Co., Ltd. (Shanghai, China). The RNA-seq dataset was uploaded to the Gene Expression Omnibus (GEO) database (GSE227623). The analysis of RNA-seq results encompassed several aspects, including the identification of differentially expressed genes, assessment of their correlation, exploration of associated pathways, potential subcellular localization, and prediction of differentially expressed proteins. Upon analysis of the sequencing data, relevant target genes were selected for further validation in subsequent steps of the study.
Quantitative real-time polymerase chain reaction (qrt-PCR)
RNA extraction was performed on kidney tissues obtained from control and model mice, as well as on HK-2 cells. Total RNA was isolated using AG RNAex Pro Reagent (AG21102, AG) and DPEC (SM-V900882, Sigma), followed by phase separation with chloroform, precipitation with isopropanol, and purification with anhydrous ethanol. Subsequently, the extracted RNA was subjected to reverse transcription using Uni All in One First Strand cDNA Synthesis SuperMix for qPCR (AU341, TransGen Biotech). The resulting cDNA was stored at −20 °C until further use. qRT-PCR was performed using SuperMix (AQ601 full gold) through standard curve method. The PCR conditions included an initial denaturation step at 95 °C for 2 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 15 s, and extension at 72 °C for 30 s. The ratio for the mRNA of interest was normalized by GAPDH expression. The specific primers used for qRT-PCR are provided in Table 1.
Primer sequence of qRT-PCR.

Transfection small interfering RNA (SiRNA)
HK-2 cells were cultured until reaching a density of 70–80% confluency, following which they were transfected with USP18-siRNA. The transfection process was carried out according to the manufacturer's instructions. Briefly, HK-2 cells were incubated with siRNA for 6 h using a transfection reagent provided by General Biol. After this incubation period, the culture medium was replaced, and LPS at a concentration of 10 μg/ml was added to the culture. Cells were then stimulated with LPS for 24 h.
siRNA oligos were synthesized by General Biol Company, and three variants USP18 siRNA were evaluated for their inhibitory efficacy. Among these variants, siRNA861 exhibited the most potent inhibition and was consequently selected for subsequent experiments. The sequence of siRNA861 was as follows: 5′-GAAGAAGACCCGUGGGAAATT-3′ (Forward) and 5′-UUUCCCACGGGUCUUCUUCTT-3′ (Reverse). Negative control sequences used were: 5′-UUCUCCGAACGUGUCACGUTT-3′ (Forward) and 5′-ACGUGACACGUUCGGAGAATT-3′ (Reverse). The knockdown efficiency of USP18-siRNA was validated through both western blot and qRT-PCR.
Western blot validation of USP18 and downstream pathway expression changes
Proteins from tissues and HK-2 cells were extracted using RIPA (P0013B, Beyotime) supplemented with protease inhibitor for 30 min at 4 °C. The lysates were then centrifuged at 12,000 rpm for 15 min at 4 °C, and the upper supernatant was collected. Protein samples were separated by 10% SDS-PAGE and transferred onto 0.45 μm PVDF membranes (Millipore, Billerica, MA). Subsequently, the membranes were blocked in 5% non-fat milk for 1h and then washed three times in TBS-Tween-20 buffer for 5min each. Next, the membranes were incubated with specific primary antibodies overnight at 4°C. After incubation with secondary antibody at 25 °C for 1 h, chemiluminescent signals were developed by ECL-chemiluminescent kit (MA0186-L, Meilunbio). Following the visualization of Actin protein (the internal control for protein loading), membranes were immersed in the first and second antibody removal solution, incubated with the target antibody again, and subjected to further development. Quantitative densitometric analyses of blotting images was performed using Image J software. Primary antibodies used were as follows: USP18 (A16739, ABclonal), PI3K (YT6156, Immunoway), AKT (YT0185, Immunoway), NF-κB P65 (YT3108, Immunoway), and actin (AC026, ABclonal). The second antibody used was goat anti-rabbit IgG antibody (Ap132p, 1:2000, Millipore).
Flow cytometry
Cell apoptosis was evaluated using the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit obtained from ?. HK2 cells, collected following siRNA transfection and LPS treatment, were resuspended in Annexin binding buffer. Subsequently, cells were double-stained with 5 μL Annexin V-FITC and 5 μL PI in the dark for 15 min. Flow cytometric analysis was performed to determine the apoptotic rate of HK2 cells using a flow cytometer.
Clinical data collection
From October 2022 to November 2022, patients diagnosed with sepsis in the Intensive Care Department of The First Affiliated Hospital of Harbin Medical University were recruited for the study. SA-AKI Patients were selected based on their levels of Cr and BUN on the first day of admission.Inclusion criteria: (1) 18 years ≤ Age ≤ 80 years; (2) Meets the sepsis 3.0 diagnostic criteria for sepsis, with elevated blood creatinine and urea nitrogen in the laboratory results, which can be diagnosed as SA-AKI. Exclusion criteria: (1) Diagnosed as a patient with sepsis, but without elevated blood creatinine and urea nitrogen, or having undergone dialysis treatment; (2) Patients with chronic kidney failure in the past; (3) Patients with underlying diseases such as tumors and immune system diseases. Blood and urine samples were collected from these SA-AKI patients, as well as from individuals with normal kidney function, for comparison. This study received approval from the Ethics Committee of The First Affiliated Hospital of Harbin Medical University (IRB-AF/SC-04/02.0).
Statistical analysis
To ensure the reliability of our findings, a power analysis was conducted, predicated on the expected differences in USP18 expression between SA-AKI models and controls, aiming for an 80% power and a 5% significance level. This analysis dictated our sample size, ensuring adequate power to detect the hypothesized effects reliably. Experiments were conducted independently three times. Statistical analysis was performed using SPSS 26.0. Quantitative data, which exhibited a normal distribution, were presented as the mean ± standard deviation (SD). Student's t-test was employed to compare differences between two groups, while one-way analysis of variance followed by Tukey's post hoc test was utilized for comparisons involving multiple groups. A p value less than 0.05 was considered statistically significant, indicating a significant difference between the compared groups (p<0.05).
Results
Verification of the SA-AKI mouse model
The intraperitoneal injection of LPS is a common method for constructing models of AKI in sepsis. After the intraperitoneal injection of 10 mg/kg LPS, mice were kept for 24 h and serum biochemical indicators and inflammatory factors were analysed. Results showed that levels of Cr, BUN, IL-6, IL-1β and TNF-α were significantly higher in the SA-AKI group than in the control group (Figure 1(a) to (e)), indicating the SA-AKI mouse model induced by intraperitoneal LPS injection was successfully established.

Successful establishment of the SA-AKI model. Mice were administered an intraperitoneal injection of 10 mg/kg LPS. (a–e) After 24 h, biochemical analysis and ELISA were performed to detect CRE, BUN, IL-6, IL-1β, TNF-α levels using respective kits. Data are presented as mean ± SD. Statistical significance denoted by *p < 0.05, **p < 0.01, ***p < 0.0001, ****p < 0.00001.
HE staining of kidney in SA-AKI mice
After successful SA-AKI modelling in mice, kidney specimens were fixed and stained with HE to determine the degree of renal glomerular injury and inflammatory infiltration in the model group. In comparison to the control group, where glomerular structures exhibited regular morphology and minimal inflammatory infiltration, the SA-AKI group displayed evident structural irregularities, such as glomerular hypertrophy, mesangial expansion, and distortion of tubular architecture. Additionally, there was a significant increase in inflammatory cell infiltration, particularly neutrophils and mononuclear cells, within the renal interstitium and glomerular tufts. These structural alterations were more pronounced at high magnification (Figure 2(a) and (b)), highlighting extensive tubular epithelial cell necrosis, loss of brush border, and interstitial edema. This indicates the presence of septic renal injury within the model group.
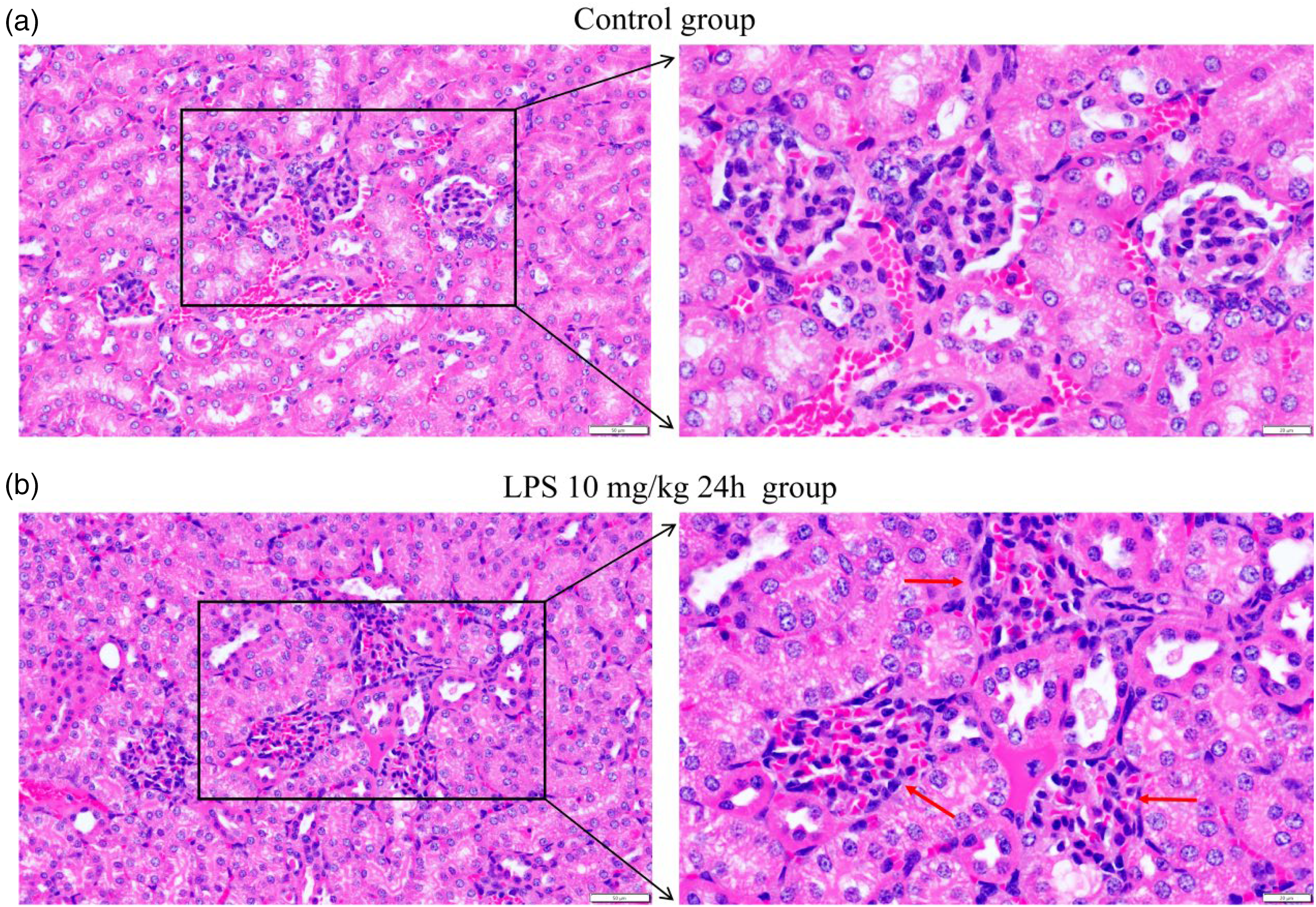
Morphological changes in kidney tissue in SA-AKI mice. HE staining was performed on kidney sections from both the control group (a) and model group (b). Representative images of renal tissue structure are shown at magnifications of 200× (left, scale bar = 50 μm), and 400× (right, scale bar = 20 μm). The red arrows indicate irregular glomerular structures with glomerular hypertrophy, mesangial expansion, and distortion of tubular architecture.
Analysis of RNA-seq-related results
After the establishment of the SA-AKI mouse model through intraperitoneal injection of LPS, 3V3 sequencing was performed. The results were analysed, and genes were selected. Between the control mice and the SA-AKI mice, there were 2672 differentially expressed genes, with 1247 upregulated genes and 1425 downregulated genes in SA-AKI mice relative to those in control mice (Figure 3(a)). Further investigation focused on the USP18 gene, which was identified as upregulated in SA-AKI mice according to the heatmap analysis. Statistical analysis confirmed significantly elevated expression of USP18 in SA-AKI mice (Figure 3(b)).

Upregulated expression of USP18 in SA-AKI mice revealed by transcriptomic analysis. (a) RNA-seq was performed on kidney samples from both the control and modeling groups (3V3). A total of 2672 differentially expressed genes were identified between the two groups. (b) Heatmap representation of the RNA-seq data.
After analysing the sequencing results for enriched pathways and functional effects, we found that the relevant differentially expressed genes were highly correlated with ubiquitin-related protease and cytokine functions, cell response to LPS, the TNF-α pathway, and apoptosis (Figure 4(a) to (e)). Therefore, we conducted experiments to further investigate inflammation-related pathways and cell apoptosis. Based on extensive literature review, we chose a classic pathway: the PI3K-AKT-NF-κB pathway for further investigation.
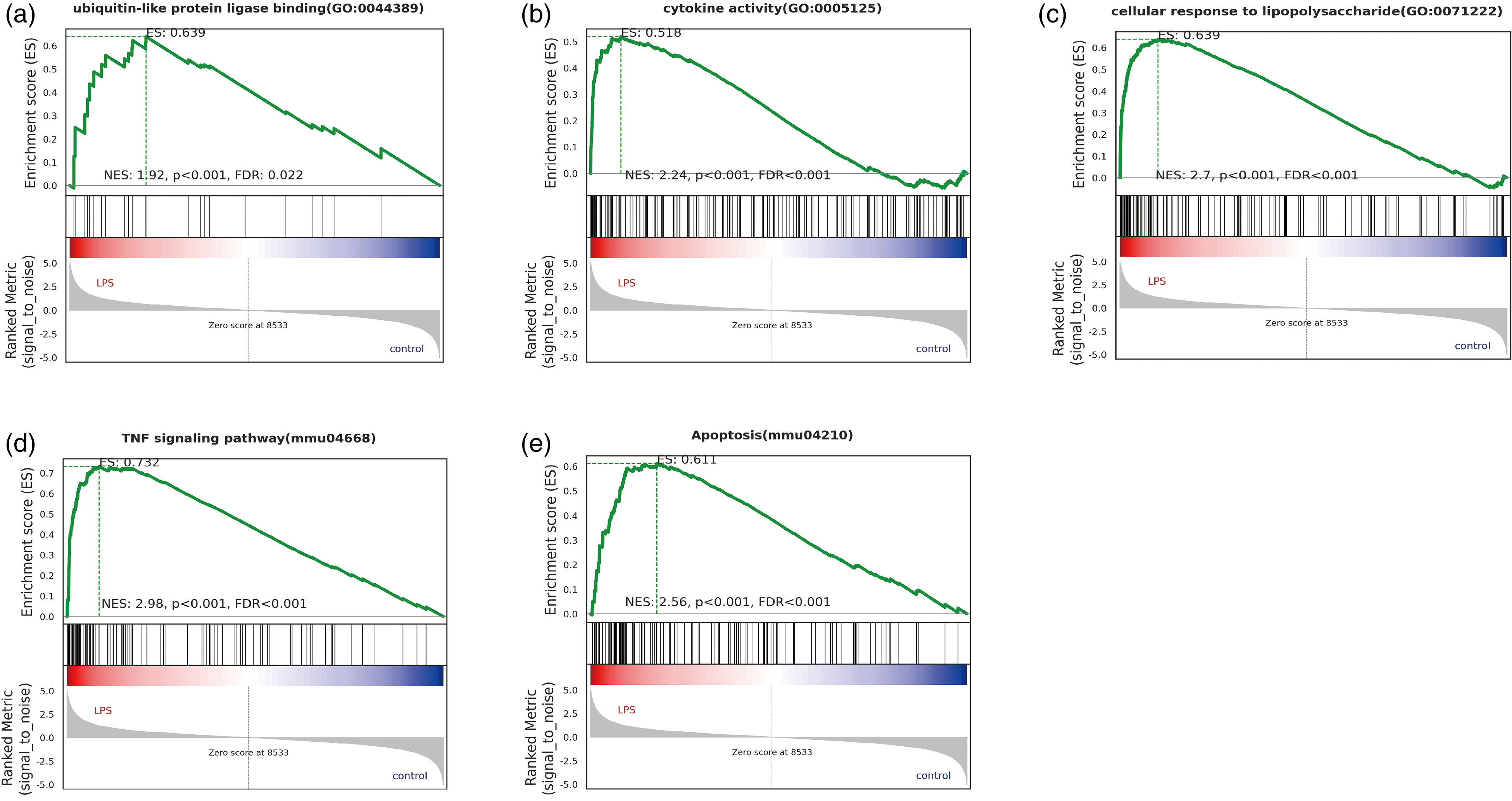
Enrichment analysis of pathways associated with differentially expressed genes. (a) Distribution of differentially expressed genes associated with ubiquitin-related proteins. (b) Differentially expressed genes related to cytokine action. (c) Genes showing differential expression in response to LPS stimulation. (d) Differential gene expression related to the TNF signaling pathway. (e) Distribution of differentially expressed genes associated with apoptosis.
Expression of USP18 and the downstream pathway in the kidneys of SA-AKI mice
Following RNA and protein extraction from the kidneys of SA-AKI mice, USP18 and key components of downstream pathways were analysed. In the kidney, the mRNA expression of USP18 was significantly upregulated (Figure 5(a)). At the protein level, there was a notable increase in USP18 expression, alongside elevated expression levels of the components within the PI3K-AKT-NF-κB pathway (Figure 5(b) to (f)). These findings underscored the involvement of USP18 and its downstream signaling cascades in the pathogenesis of SA-AKI.
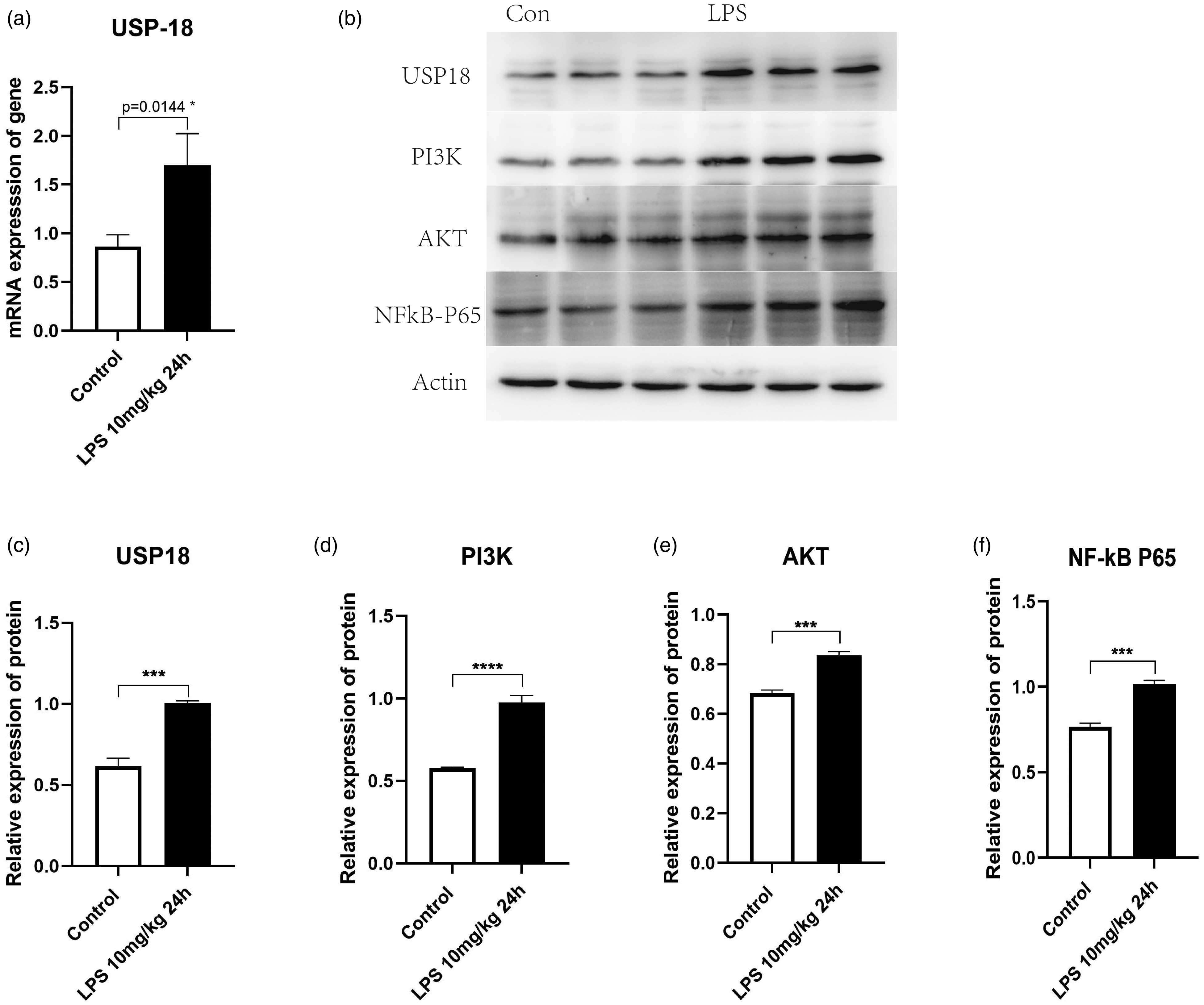
Upregulated expression of USP18 and the PI3K-AKT-NF-κB pathway in the kidneys of SA-AKI mice. (a–f) Western blot analysis was employed to assess the expression levels of USP18 and proteins associated with the PI3K-AKT-NF-κB pathway in both the control and model groups. Data are presented as mean ± SD, and experiments were conducted with biological triplicates. Statistical significance is indicated by *p < 0.05, ***p < 0.001, ****p < 0.0001.
mRNA levels of IL-6, TNF-α and USP18 in LPS-stimulated HK-2 cells
HK-2 cells are human renal tubular epithelial cells, and treatment with LPS can cause corresponding inflammatory damage to the cells. After stimulating HK-2 cells with different concentrations of LPS (1 µg/ml, 5 µg/ml, and 10 µg/ml), the expression of IL-6 and TNF-α was assessed at the mRNA level by qRT-PCR. The results showed that in the HK-2 cell model constructed with 10 µg/ml LPS, the expression of IL-6 and TNF-α was significantly upregulated (Figure 6(a) and (b)), indicative of a pronounced inflammatory response. Based on these results, we utilized 10 µg/ml LPS to establish an LPS-induced SA-AKI model in HK-2 cells.

Increased mRNA levels of IL-6, TNF-α, and USP18 in LPS-stimulated HK-2 cells. (a, b) qRT-PCR analysis was performed to assess IL-6 and TNF-α mRNA levels in HK-2 cells stimulated with 1, 5, and 10 μg/ml LPS. (c) qRT-PCR analysis of USP18 mRNA levels in HK-2 cells treated with 10 μg/ml LPS for 24h compared to the control group. (d) HK-2 cells were transfected with three different USP18 siRNAs, and qRT-PCR was used to measure USP18 mRNA levels. siRNA861 exhibited the highest inhibitory effect. Data are presented as mean ± SD, and experiments were conducted with biological triplicates. Statistical significance denoted by *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
After treating HK-2 cells with LPS, mRNA was extracted to assess USP18 expression. USP18 expression was significantly upregulated in the LPS model (Figure 6(c)). To investigate the functional role of USP18, we employed siRNA to knock down its expression. The results demonstrated significant knockdown of USP18 expression with siRNA treatment (Figure 6(d)). Among the tested siRNAs, siRNA861 exhibited the most substantial reduction in USP18 expression and was thus selected for subsequent experiments.
Inflammatory response changes in HK-2 cells treated with LPS and USP18 siRNA
After using siRNA861 to knock down the expression of USP18 in HK-2 cells, the cells were treated with LPS for 24 h. The mRNA and protein in HK-2 cells (control group, LPS group, and siRNA group) were extracted to analyse related inflammatory factors. After knocking down USP18, the expression of IL-6 and TNF-α was upregulated (Figure 7(a) and (b)), and the expression levels of components of the PI3K-AKT-NF-κB inflammatory response pathway increased (Figure 7(c) to (g)).

Changes in inflammatory response in HK-2 cells treated with LPS and USP18 siRNA. (a, b) qRT-PCR analysis of IL-6 and TNF-α mRNA levels in the control group, LPS-treated group (10 μg/ml for 24 h), and LPS + siRNA-treated group (10 μg/ml LPS + siRNA for 24 h). (c–g) Western blot analysis of the expression levels of USP18, PI3K, AKT, and NF-κB P65 in the same experimental groups. Experiments were conducted with biological triplicates. Statistical significance denoted by *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
Changes in HK-2 cell apoptosis after treatment with LPS infection and USP18 siRNA
After knocking down USP18 and treating HK-2 cells with LPS for 24 h, flow cytometry was performed to detect HK-2 cell apoptosis. The rate of early apoptosis of HK-2 cells significantly increased after 24 h of LPS stimulation, but after knocking down USP18 expression, the rate of early apoptosis of HK-2 cells significantly decreased (Figure 8(a) to (m)).

Knockdown of USP18 was found to influence the proportion of early apoptosis in HK-2 cells. Flow cytometry was utilized to assess apoptosis in HK-2 cells following infection with LPS at 10 μg/ml with or without USP18 knockdown in with siRNA. Early apoptosis was detected using flow-based single dye detection methods (a–c). The proportion of apoptosis in HK-2 cells was evaluated in the control group (d–f), following LPS infection (g–i), and following LPS infection with USP18 knockdown (j–l). Statistical analysis of the proportion of early apoptosis in the second quadrant is presented in panel (m). Statistical significance denoted by *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
Changes in USP18 expression in the blood and urine of patients with sepsis and AKI
The expression of USP18 in the blood and urine of patients with SA-AKI was measured by ELISA and compared with that in the blood and urine of healthy individuals. These patients were diagnosed with sepsis (Sepsis 3.0) and had elevated levels of Cr and BUN (Figure 9(a) and (b)). Analysis of blood and urine samples from patients with SA-AKI revealed higher levels of USP18 expression compared to samples obtained from healthy individuals (Figure 9(c) and (d)).

Increased expression of USP18 in blood and urine in SA-AKI patients. (a, b) Comparison of blood Cr and BUN levels between normal person individuals and SA-AKI patients (n = 20). (c, d) Detection of USP18 expression in the serum and urine of SA-AKI patients and normal individuals using ELISA. Statistical significance denoted by *p < 0.01, **p < 0.05, ***p < 0.001, ****p < 0.0001.
Discussion
SA-AKI is a significant cause of morbidity and mortality in critically ill patients, characterized by systemic inflammation and organ dysfunction.1,2 To model SA-AKI in mice, we selected a 10 mg/kg dosage of LPS for intraperitoneal injection, a choice well-supported by extensive prior research demonstrating its efficacy in inducing sepsis and acute kidney injury, making it one of the most commonly used inducers of SA-AKI.22,23 After establishing a mouse model of SA-AKI, RNA sequencing was conducted to explore potential characteristic markers. Through analysis of gene functions and interacting proteins, USP18 emerged as a promising candidate. Subsequent analyses revealed that USP18 expression was significantly upregulated in the kidneys of SA-AKI mice, modulating immune responses, inflammatory pathways, and cell apoptosis, which may be correlated with increased activation of the PI3K-AKT-NF-κB pathway. These findings suggest that USP18 could serve as a potential biomarker and therapeutic target for early diagnosis and treatment of SA-AKI.
Gene set enrichment analysis (GSEA) was conducted to identify pathway associated differentially expressed genes. Notably, these genes were predominately enriched in immune response processes and inflammatory response functions in biological processes, cellular functions, and molecular mechanisms. However, validating the functional significance of multiple differentially expressed genes can be resource-intensive in terms of time, cost, and experimental capacity. USP18 is known to play diverse roles, including modulation of immune responses, inflammatory responses, and cell apoptosis.24–26 Previous research indicated a strong association between SA-AKI and immune-inflammatory mechanisms.27–30 A Study have linked USP18 deficiency to heightened sensitivity to LPS-induced lethality, highlighting the role of TRIF-dependent pathway in protein ISGylation and USP18 expression upon LPS treatment. 31 Given the pivotal role of USP18 in inflammation and immune cell pathways, it was selected as the target gene for our investigation into SA-AKI. This focused approach allows for more efficient use of resources while providing significant insights into the pathogenesis of SA-AKI. Furthermore, pathway analyses highlighted significant enrichment in inflammatory pathways such as the NF-κB pathway and cytokine response pathway. Therefore, our focus turned towards investigating the impact of USP18 on downstream pathways, particularly those pertinent to inflammation. Following a comprehensive review of existing literature, we identified the PI3K-AKT-NF-κB inflammatory pathway as a likely target regulated by USP18 in SA-AKI.
Our GSEA results also revealed a correlation between the biological function of differentially expressed genes and immune response as well as cell apoptosis. We utilized HK-2 cells, an immortalized cell line derived from the normal adult human kidney, to simulate physiological conditions in vitro. HK-2 cells have been extensively used in various kidney disease models, particularly acute kidney injury, due to their established characteristics and reliability in studying renal pathophysiology.32,33 Our findings indicated a correlation between USP18 and tumor cell apoptosis. Moreover, experimentation in an HK-2 cell model demonstrated that USP18 can promote early cell apoptosis. Considering that sepsis-induced kidney damage primarily arises from immune reactions or exacerbated inflammatory reactions, 34 we pursued further investigations into the expression of USP18 in SA-AKI and the resultant changes in inflammatory responses.
The western blot and qRT-PCR results indicated that the protein expression and mRNA of USP18 was indeed upregulated in the kidneys of SA-AKI mice. This finding consistent with observations in patient samples. To corroborate these findings clinically, we compared USP18 expression levels in blood and urine samples obtained from patients with SA-AKI and healthy individuals, revealing significantly higher expression levels in the former group. In addition, our examination of the classic PI3K-AKT-NF-κB inflammatory pathway unveiled heightened expression levels of essential molecules within this cascade in the kidneys, 35 further underscoring the significance of this pathway in our study. Subsequent validation in a SA-AKI mouse model reinforced our observations. To simulate cellular injury, HK-2 cells were treated with LPS to establish a cell injury model. In this model, both USP18 expression and the expression of components within the PI3K-AKT-NF-κB pathway were markedly increased, providing additional support for our hypothesis.
After we confirming the upregulation of USP18 in SA-AKI, we used siRNA to knock down the expression of USP18 in HK-2 cells. In the HK-2 cell model, the downregulation of USP18 resulted in increased expression levels of downstream components within the PI3K-AKT-NF-κB inflammatory pathway, alongside elevated expression of IL-6 and TNF-α. This observation suggested that USP18 plays a regulatory role in the inflammatory response by inhibiting the PI3K-AKT-NF-κB pathway.
Besides, USP18 has been implicated in the regulation of cell apoptosis. Previous studies in tumorigenesis have demonstrated correlation between USP18 expression and both tumour cell proliferation and apoptosis.36,37 Notably, NF-κB, known for its anti-apoptotic function, is regulated by USP18. Therefore, we hypothesized that USP18 may influence apoptosis in HK-2 cells. Upon downregulating USP18, we observed a decrease in the apoptosis rate of cells. Specifically, in the HK-2 cell model of SA-AKI, knocking down USP18 expression inhibited the early apoptosis in injured HK-2 cells. These findings collectively suggest that USP18 exerts a certain level of influence on the early apoptosis of injured HK-2 cells.
Several limitations exist within this study. Firstly, our assessment was confined to observing the relevant changes subsequent to USP18 knockdown solely within cells. Unfortunately, we did not identify suitable drugs to inhibit USP18 gene expression in mice. Moreover, the inclusion of USP18 knockout mice is imperative for comprehensive validation. Secondly, while we meticulously analysed inflammatory responses and early apoptosis changes following USP18 knockdown in cells, we neglected to investigate the corresponding alterations upon USP18 overexpression. Furthermore, the impact of USP18 on inflammation under normal physiological conditions remains unexplored within our study. Thus, further investigations are warranted to elucidate the causal relationship between USP18 and inflammatory responses. Lastly, the sample size for applying USP18 as a biomarker at the clinical level was insufficient. Future research should incorporate proteomics and other relevant studies to address this gap adequately.
Conclusion
Our study revealed an upregulation of USP18 expression in SA-AKI, where it exerted inhibitory effects on the PI3K-AKT-NF-κB pathway, thereby modulating inflammatory response. Additionally, USP18 demonstrated a notable influence on the apoptosis of epithelial cells in SA-AKI. Importantly, elevated levels of USP18 were detected in the blood and urine of clinical patients. These findings suggested USP18's potential as a biomarker for SA-AKI, with promising prospects for future diagnostic applications. Further investigations into the utility of USP18 in clinical settings hold significant promise for enhancing diagnostic accuracy and improving patient outcomes.
Footnotes
Acknowledgements
Not applicable.
Author contributions
YC and MZ conceptualized and designed the study, drafted the initial manuscript. YW, QG, JZ, XW, XM, YL, WY, NL, HL and QG collected the data and carried out the initial analyses. MZ critically reviewed the manuscript for important intellectual content. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Availability of data and materials
All data are incorporated into the article. The data underlying this article are available in the article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Compliance with ethical standards and has passed clinical and animal ethics. The Ethics Committee of the First Affiliated Hospital of Harbin Medical University passed the clinical and animal ethics. The operation of this experiment is in line with the principles of laboratory animal management and welfare ethics. The experiment is conducted for clinical patients according to the principle of no harm. The ethical codes are: IACUC: 2022080 and IRB-AF/SC-04/02.0 respectively. The ethics have already showed that they agree to consent to participate and consent to publish.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China, Heilongjiang Province Key R&D Program, The experiments were done in Heilongjiang Province Key Laboratory of Critical Care Medicine, (grant number 82172164, GA21C011, JD22C005).