
Abstract
Objective
Excessive inflammatory responses in the endocardium are related to progression of infectious endocarditis. This study aimed to investigate whether (Z)-7,4'-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside (DHAG), a compound isolated from the endophytic fungus Penicillium citrinum of Bruguiera gymnorrhiza, could attenuate cell damage caused by lipoteichoic acid (LTA) in embryonic rat heart cells (H9c2).
Methods
LTA-induced cell damage occurred in H9c2 cells and the protective effects of DHAG at different concentrations (1–10 µM) were assessed. Indicators of oxidative stress and inflammatory responses in H9c2 cells were measured.
Results
DHAG (1–10 µM) significantly attenuated LTA-induced damage in H9c2 cells, as evidenced by increased cell viability and mitochondrial membrane potential, decreased cytochrome c release and DNA fragmentation, inhibition of caspase-3 and -9 activity, and altered expression of apoptosis-related proteins. DHAG also decreased oxidative stress by increasing protein expression of nuclear factor (erythroid-derived 2)-like 2 (Nrf2). Furthermore, DHAG inhibited inflammatory responses by decreasing protein expression of nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs).
Conclusion
DHAG exerted protective effects against LTA-induced cell damage, at least partially by decreasing oxidative stress and inhibiting inflammatory responses. Our results provide a scientific rational for developing DHAG as a therapy against infectious endocarditis.
Keywords
Introduction
Infective endocarditis (IE) results from infection of the endocardium by bacteria (commonly Streptococcus sanguinis) or fungi, and may cause cardiac complications such as fever, petechiae, anemia, and valvular incompetence or obstruction.1,2 Unfortunately, little is known regarding the molecular mechanisms causing IE.3,4 Current treatments include intravenous antibiotics, valve debridement, valve repair or replacement and surgery. However, few effective therapies are available for patients with IE.2,5,6 Therefore, the search for novel therapeutic strategies is critically important.
As the smallest bioactive peptidoglycan fragment present in the membranes of Gram-positive bacteria, lipoteichoic acid (LTA) is a major antigen recognized by innate immune cells such as macrophages and monocytes and is well-known for its induction of inflammatory responses.7,8 Studies showed that LTA triggered activation of cardiomyocytes and induced a diverse array of inflammatory responses by promoting the release of various pro-inflammatory cytokines and mediators, including nitric oxide (NO), interleukin (IL)-1β, IL-6, and tumor necrosis factor alpha (TNF-α) through the phosphorylation of nuclear factor-κB (NF-κB). 9 Over-production of pro-inflammatory factors resulted in toxic intracellular events, resulting in cell death. 10 Excessive amount of pro-inflammatory cytokines can be detected in IE and these cytokines stimulate oxidant production in the myocardium with subsequent peroxidative damage to macromolecules. Oxidative stress has been shown to play a crucial role in the IE progression. 11
Herbal plants are an important source of medicinal products. Supplementation of exogenous antioxidants might alleviate oxidative damage and different antioxidants have been used to effectively treat disorders caused by oxidative stress.12–14 (Z)-7,4'-dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside (DHAG), a compound isolated from the endophytic fungus Penicillium citrinum of Bruguiera gymnorrhiza., was shown to have neuroprotective effects via inhibition of oxidative stress. 15 Furthermore, DHAG was also found to inhibit LTA-induced production of inflammatory factors in our preliminary study. In the present study, the protective effects of DHAG against LTA-induced inflammatory responses and oxidative stress in cardiomyoblasts were investigated.
Materials and methods
Cell lines, reagents and chemicals
The cardiomyoblast cell line H9c2 was purchased from Beijing Cell bank (Beijing, China). Fetal bovine serum and RPMI-1640 medium were from Gibco BRL (Gaithersburg, MD, USA). LTA and 3-(4,5-dimethylthiazol)-2,5-diphenyltetrazolium-bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Caspase-3/9 activity assay kits were from Promega (Madison, WI, USA). Glutathione (GSH), superoxide dismutase (SOD), malondialdehyde (MDA), IL-1β, and TNF-α assay kits were purchased from Jiancheng Biological Engineering (Nanjing, China). The cytochrome c assay kit was from R&D systems (Minneapolis, MN, USA). 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) and 2′,7′-dichlorofluorescin diacetate (DCFH-DA) were purchased from Molecular Probes (Eugene, OR, USA). Real-time PCR reagents were purchased from Thermo Fisher (Waltham, MA, USA). DHAG was provided by Prof. Teng from Zhengzhou University. 15 All solvents and chemicals were analytical grade and were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). LTA and DHAG were dissolved in dimethyl sulfoxide (DMSO).
Cell culture and treatment
H9c2 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 1% streptomycin/penicillin at 37°C under a humidified atmosphere containing 5% CO2. H9c2 cells were treated with DHAG at 1, 3, or 10 µM for 6 hours, then with LTA at 10 µg/mL for 24 hours. Cells were treated with DMSO as a negative control.
Cell viability assay
Cytotoxicity was assessed using an MTT-based colorimetric assay. After treatment, MTT solution was added to each well to a final concentration of 62.5 µg/mL and mixed well. After incubation for 4 hours at 37°C, the supernatant was removed and the formazan produced in viable cells was solubilized with 200 µL of DMSO. The absorbance in each well was measured at 570 nm using an enzyme-linked immunosorbent assay (ELISA) microplate reader.
Mitochondrial membrane potential (MMP) measurement
MMP was measured using the fluorescent probe JC-1. After treatment, H9c2 cells were incubated with 2 µM JC-1 at 37°C for 15 min in the dark. After washing, fluorescence was measured using a fluorescence reader with excitation at 490 nm and emission at 590/530 nm.
Cytochrome c measurement
H9c2 cells were fractionated after treatment. Levels of cytochrome c were measured using an assay kit according to the manufacturer’s instructions. Optical density was measured at 490 nm using an ELISA plate reader.
DNA fragmentation measurement
After treatment, H9c2 cells were lysed and DNA fragmentation was measured using a Cell Death Detection ELISAplus kit according to the manufacturer’s instructions. The optical density was measured at 405 nm using a microplate reader.
Caspase activity measurement
Caspase-3/9 activities were measured according to the manufacture’s protocol. Briefly, 100 µL of reagent for specific caspase were added into each well and the plate was then incubated at room temperature for 3 hours. Luminescence was measured using a microplate reader.
SOD activity, MDA and GSH level
SOD activity and MDA and GSH levels were measured using assay kits according to the manufacturer’s protocols. Briefly, after washing, H9c2 cells were homogenized and the supernatant was used to measure SOD, MDA and GSH.
Measurement of reactive oxygen species (ROS)
Intracellular ROS were measured using the fluoroprobe DCFH-DA. H9c2 cells were incubated with DCFH-DA for 30 min in the dark. After washing twice, fluorescence intensity was measured at 488/525 nm using a fluorescence microplate reader.
Cytokine measurement
After treatment, supernatants of culture medium were collected. Levels of IL-1β and TNF-α were quantified using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions.
NO measurement
NO was measured using a Griess assay (Sigma-Aldrich). 16
Real-time RT-PCR
Total RNA was extracted from H9c2 cells using 1 mL of TRIzol reagent. RNA (0.5 µg) was reverse transcribed into cDNA using a PrimeScript™ RT reagent kit (Takara, Japan). Real-time RT-PCR was performed on a StepOne system (Thermo Fisher) using SYBR green Supermix (Thermo Fisher) and the comparative Ct value method. Expression data were normalized to the geometric mean of the β-actin housekeeping gene. The gene-specific primer sequences used were as follows: iNOS, forward: agggagtgttgttccaggtg, reverse: tcctcaacctgctcctcact, NCBI reference: NM_012611.3; IL-1β, forward: agtctgcacagttccccaac, reverse: agacctgacttggcagagga, NCBI reference: NM_031512.2; TNFα, forward: gaaacacacgagacgctgaa, reverse: cagtctgggaagctctgagg, NCBI reference: NM_012675.3.; and β-actin, forward: gtcgtaccactggcattgtg, reverse: tctcagctgtggtggtgaag, NCBI reference: NM_031144.3.
Western blotting
H9c2 cells were harvested in protein lysis buffer containing protease inhibitors and phosphatase inhibitors. Total protein content was determined using Bradford reagent (Beyotime, Shanghai, China). Equal amounts of protein (45 µ g) were mixed with 4× loading buffer and separated in 12% (w/v) SDS-polyacrylamide gels. Proteins were then transferred onto polyvinylidene fluoride membranes. Membranes were blocked in Tris-buffered saline containing 1% bovine serum albumin and 0.1% Tween-20 for 1 hour at room temperature. The membranes were incubated with primary antibodies [anti-cleaved caspase-3 rabbit polyclonal (p)Ab ab49822, 1:1000; anti-cleaved caspase-9 (Asp353) rabbit pAb #9507, 1:1000; anti-Bcl-2 rabbit pAb ab196495, 1:1000; anti-Bax rabbit monoclonal (m)Ab ab32503, 1:1000; anti-NADPH oxidase 4 rabbit pAb ab154244, 1:1000; anti-HO-1 rabbit pAb ab13243, 1:1000; anti-Nrf2 rabbit pAb ab137550, 1:1500; anti-P65 rabbit pAb ab16502, 1:1000; anti-P65 (phosphor-S536) rabbit pAb ab86299, 1:1000; anti-P38 rabbit mAb, ab170099, 1:1000; anti-P38 (phosphor-Y182) rabbit pAb, ab47363, 1:1000; anti-JNK rabbit mAb, ab179461, 1:1000; anti-JNK (phospho-T183+T183+T221) rabbit mAb, ab124956, 1:1000; and anti-β-actin, rabbit pAb, ab16039,1:3000] at 4°C overnight. After washing, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-rabbit IgG H&L HRP, ab6721, 1:3000) for 1 hour at room temperature. All antibodies were from Abcam (Cambridge UK) except for anti-cleaved caspase-9 (Asp353) rabbit pAb, which was purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody binding was visualized using an enhanced chemiluminescence system (Millipore, Billerica, MA, USA).
Statistical analysis
Data were expressed as mean ± standard deviations and analyzed with SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). The Kolmogorov-Smirnov test was used to assess normality. Dunnett’s t test was used to compare the experimental group with the control group. Differences between two groups were compared using the t-test. Differences among multiple groups were compared using one-way analysis of variance. Values of p < 0.05 were considered statistically significant. Samples were measured in triplicate and each experiment was repeated three times.
Results
DHAG treatment had no effect on H9c2 cell viability
H9c2 cells were treated with DHAG at 1, 3, or 10 µ M for 48 hours. No obvious changes in cell viability were observed by MTT assay compared with negative control cells, suggesting that DHAG treatment at 10 µM did not damage H9c2 cells (Figure 1).

H9c2 cells were treated with DHAG at 1, 3, or 10 µM for 48 hours and cell viability was assessed using an MTT assay. DHAG caused no obvious cytotoxicity in H9c2 cells.
DHAG treatment attenuated LTA-induced H9c2 cell damage
LTA was toxic to H9c2 cells. However, pre-treatment with DHAG increased cell viability (Figure 2a), increased MMP (Figure 2b), decreased cytochrome c release (Figure 2c), decreased DNA fragmentation (Figure 2d), decreased caspase activities (Figure 2e), and changed the expression levels of apoptosis-related proteins (Figure 2f).
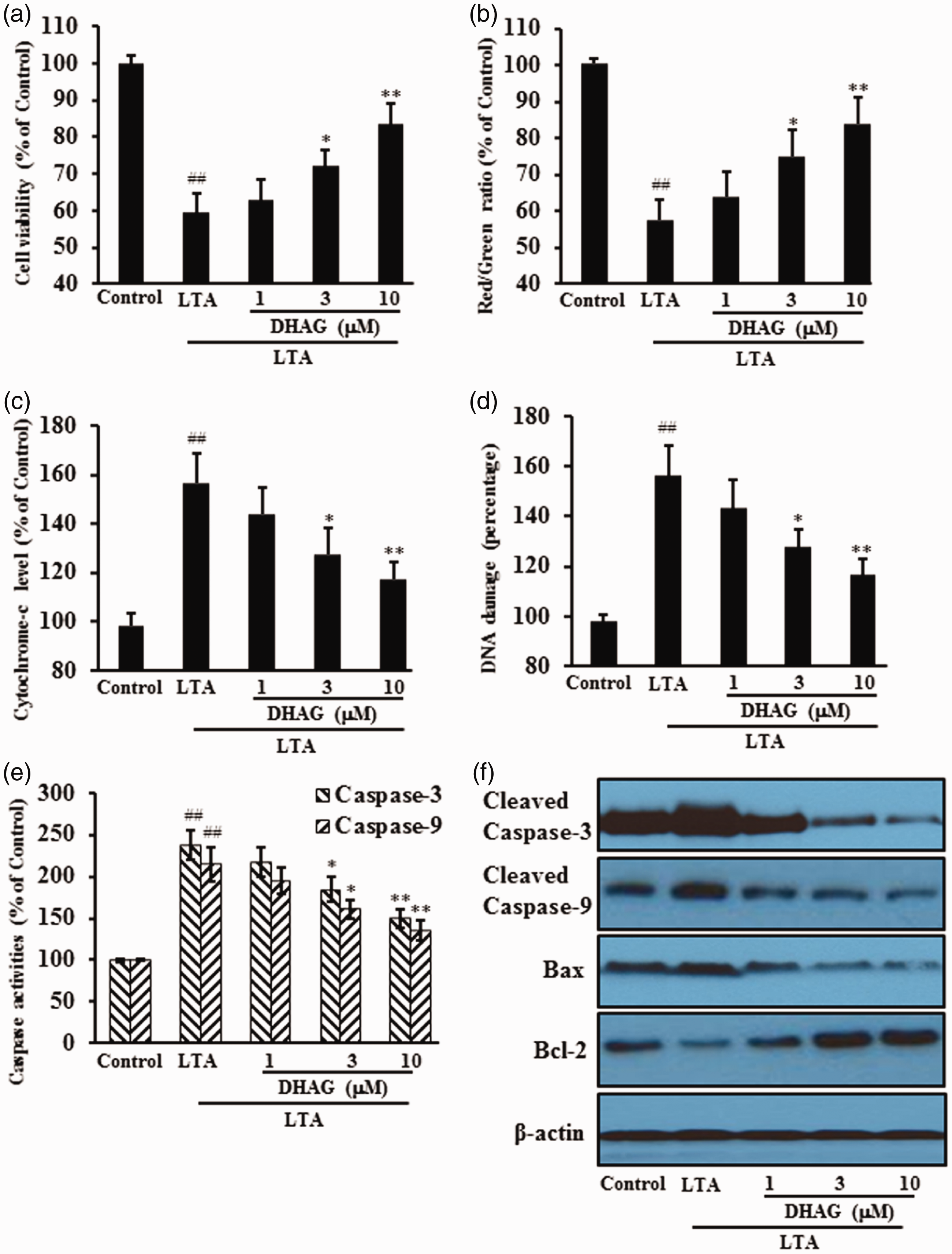
DHAG protected H9c2 cells against LTA-induced cell damage. H9c2 cells were treated with DHAG at 1, 3, or 10 µM for 6 hours then with LTA for 24 hours. Cells were collected to measure cytotoxicity via cell viability (a), mitochondrial membrane potential (b), cytochrome c release (c), DNA damage (d), and caspase activities (e). Expression of apoptosis-related proteins was also assessed by western blotting (f). ##p < 0.01 vs. negative control; *p < 0.05, **p < 0.01 vs. LTA alone.
DHAG treatment decreased oxidative stress
Oxidative stress significantly increased following LTA treatment. However, pre-treatment with DHAG significantly reduced production of ROS and MDA and increased GSH levels and SOD activities. Moreover, DHAG pre-treatment decreased NADPH oxidase 4 protein expression and increased protein expression of nuclear Nrf2 and HO-1, indicating that DHAG treatment effectively inhibited oxidative stress induced by LTA (Figure 3).

DHAG inhibited oxidative stress in H9c2 cells induced by LTA. H9c2 cells were treated with DHAG at 1, 3, or 10 µM for 6 hours and then treated with LTA for 24 hours. Cells were collected to measure oxidative stress via MDA content (a), intracellular ROS levels (b), SOD activity (c), GSH levels (d) and oxidative stress-related protein expression levels (e). ##p < 0.01 vs. negative control; *p < 0.05, **p < 0.01 vs. LTA alone.
DHAG treatment inhibited inflammatory responses in H9c2 cells
Pro-inflammatory mediators are also inflammatory markers and are present in activated cells. DHAG significant inhibited production of NO, IL-1β and TNF-α in LTA-stimulated H9c2 cells. Levels of iNOS, IL-1β and TNF-α mRNA also increased following exposure to LTA, but decreased following DHAG treatment. Moreover, levels of p-p65, p-JNK and p-p38 proteins increased following LTA stimulation, but decreased following DHAG treatment in a dose-dependent manner (Figure 4).
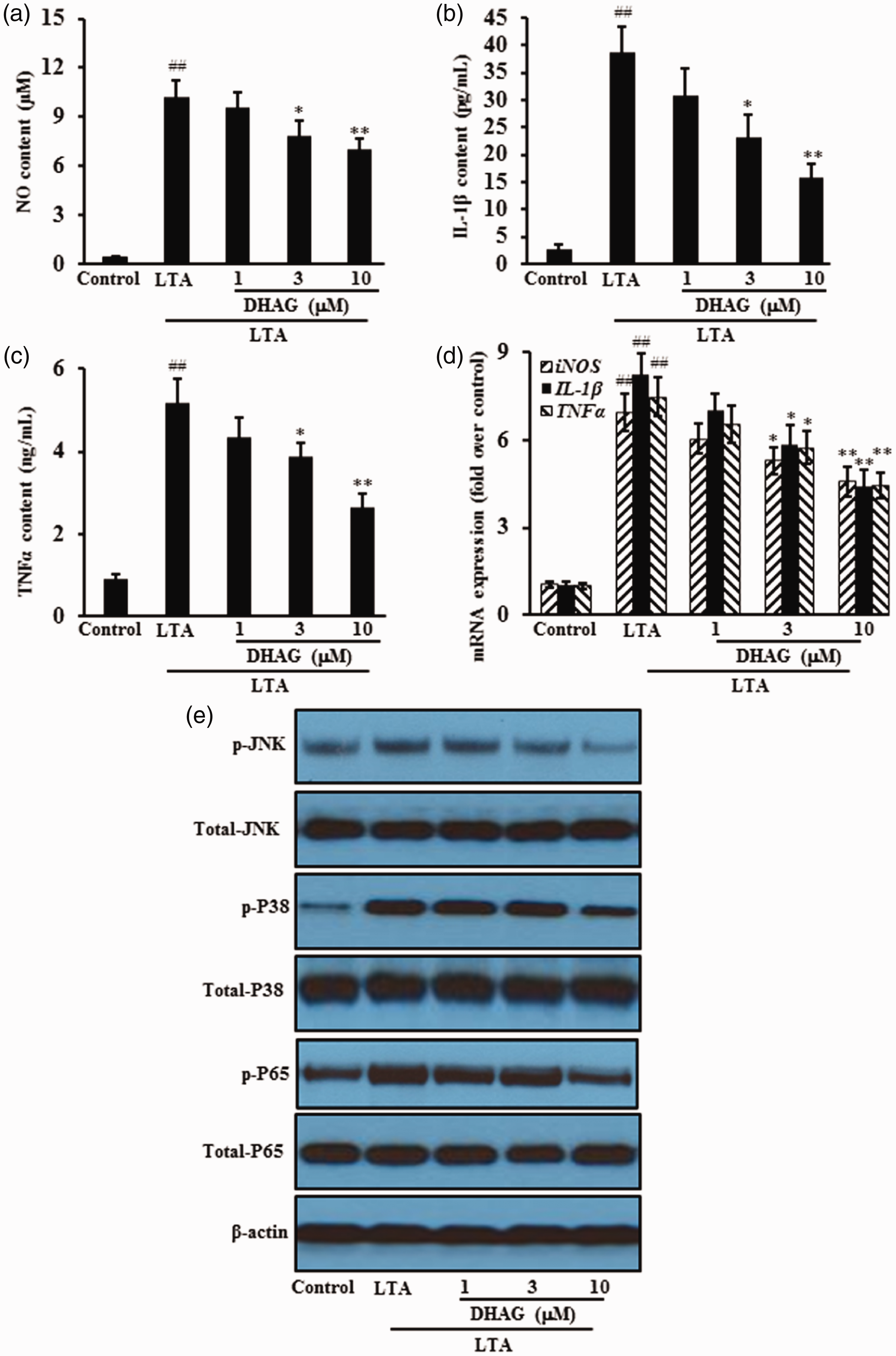
DHAG inhibited inflammatory responses in H9c2 cells induced by LTA. H9c2 cells were treated with DHAG at 1, 3, or 10 µM for 6 hours and then treated with LTA for 24 hours. Inflammatory responses were assessed via NO levels (a); IL-1β levels (b); TNF-α levels (c); mRNA expression (d) and protein expression (e). ##p < 0.01 vs. negative control; *p < 0.05, **p < 0.01 vs. LTA alone.
Discussion
The results of our study demonstrated that DHAG, a novel antioxidant, induced expression of Nrf2, which appeared to be involved in intracellular defense against LTA-mediated inflammatory responses. DHAG decreased oxidative stress and production of pro-inflammatory cytokines. Furthermore, DHAG suppressed LTA-mediated activation of upstream signaling molecules, including NF-κB and MAPKs, involved in inflammatory responses. Cardiomyocytes produce pro-inflammatory cytokines and chronic inflammatory responses can cause oxidative stress, resulting in IE.17–19 Our results revealed that DHAG attenuated the toxicity of a bacterial component and had anti-inflammatory effects that stimulated cardiomyocytes.
Mitochondria play a pivotal role in the regulation of cell death. Decreased MMP causes mitochondrial fragmentation, generating excessive amounts of ROS and causing damage to mitochondrial structure and function. Eventually, the cellular ATP supply is interrupted and apoptosis is activated.20,21 Mitochondria modulate cell viability through ROS-mediated cell oxidative injury and HtrA2/Omi liberation-induced caspase activation.22,23 As apoptosis effectors, caspase-3 and caspase-9 precursor activate endonucleases to cleave nuclear DNA, leading to cell death. 24 In this study, LTA treatment caused cell damage as shown by MMP decrease, cytochrome c release, and increased caspase-3 and -9 activities. Anti-apoptotic protein expression decreased, but pro-apoptotic protein expression increased. However, the above effects were largely reversed by DHAG treatment.
Excessive accumulation of ROS can result in oxidative stress. High oxidative stress can induce different cell death programs, including apoptosis, autophagy and necrosis,25–28 and has been considered as a major cause of cellular injuries in many disorders.29–31 MDA is well known as a widely-used marker for oxidative damage and resulting thiobarbituric acid reactive substances, whose levels are proportional to the degree of lipid peroxidation and oxidative stress. 32 Therefore, it seems reasonable to use exogenous antioxidants as potential therapeutics to reverse the imbalance between the intracellular oxidative and anti-oxidative systems. The transcription factor Nrf2 is a master regulator of antioxidant and detoxification genes with cytoprotective function. 33 Since Nrf2 inactivation is necessary for the complete execution of apoptosis in the presence of cellular damage caused by oxidative stress, constitutive activation of Nrf2 may protect cells from apoptosis via binding to antioxidant response elements and inducing the expression cytoprotective target proteins such as antioxidant proteins, phase II detoxifying enzymes, and molecular proteasome/chaperones.34,35 In the present study, DHAG-induced Nrf2 activation was observed. This further induced expression of antioxidant genes to restore oxidative homeostasis, as shown by decreased protein expression of NADPH oxidase 4,a major enzyme responsible for production of superoxide by transferring electrons across the membrane from NAD(P)H to molecular oxygen, 36 reduced levels of MDA and ROS, increased SOD activity and increased GSH levels.
Production of inflammatory cytokines is part of the immune response to many inflammatory stimuli and is involved in the progression of inflammatory diseases.37,38 DHAG exerted anti-inflammatory effects by inhibiting inflammatory factors including the pro-inflammatory mediator NO and the pro-inflammatory cytokines IL-1β and TNF-α in LTA-stimulated H9c2 cells. Moreover, DHAG considerably suppressed pro-inflammatory molecule production from upstream signaling pathways, which were involved in the progression of inflammatory responses in H9c2 cells. Activation of NF-κB and MAPK leads to transcription factor binding to the promoter regions of pro-inflammatory cytokine genes, thereby enabling transduction of extracellular signals into cellular reactions. 39 DHAG decreased phosphorylation of several MAPKs, including JNK and P38, whose phosphorylation was induced by LTA stimulation. In addition, DHAG reduced nuclear translocation of NF-κB in response to LTA.
Our study demonstrated that DHAG, isolated from the endophytic fungus P. citrinum of B. gymnorrhiza., inhibited LTA-induced oxidative stress and inflammatory responses in cardiomyoblasts. Our results provide a scientific rationale for developing DHAG as a therapeutic agent infective endocarditis.
Footnotes
Acknowledgement
This study was supported by the National Natural Science Foundation of China (Grant No. 81800052).
Consent
Patient consent was not applicable as animal and human subjects were not involved in this study.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Ethical approval
Ethical approval was not sought as animal and human subjects were not involved in this study.
Funding
This study was supported by the National Natural Science Foundation of China (Grant No. 81800052).