
Abstract
Aim:
It has been reported that angiotensin converting enzyme 2 (ACE2) is an endogenous counter-regulator of the renin–angiotensin–aldosterone system. However, angiotensin converting enzyme (ACE)/ACE2 balance in the development of human heart failure is not well established.
Methods:
Here we evaluated the expression of ACE and ACE2 at the mRNA and protein levels in the myocardium of 78 patients with mild or moderate to severe heart failure and in 13 cases with normal myocardium.
Results:
In the myocardium of patients with dilated or ischemic cardiomyopathy, ACE and ACE2 expression at the mRNA and protein levels was significantly increased compared with those in normal myocardium (P<0.01, P<0.01, respectively). The ratios of ACE/ACE2 mRNA and ACE/ACE2 were lower in the myocardium of patients with mild heart failure than those in normal myocardium but higher than those in patients with moderate to severe heart failure.
Conclusions:
ACE and ACE2 expression at the mRNA and protein levels are significantly increased in the myocardium of patients with heart failure. The compensatory mechanism of patients with mild heart may cause the decreased ACE/ACE2 ratio. However, increased ACE/ACE2 ratios may induce angiotensin II over-activation and accelerate cardiac remodeling in patients with moderate to severe heart failure.
Keywords
Introduction
The deleterious effects of the renin–angiotensin–aldosterone system (RAAS) are primarily due to increased production of angiotensin II (Ang II), which results in cardiac remodeling and heart failure. Angiotensin converting enzyme 2 (ACE2), a homolog of angiotensin converting enzyme (ACE), was recently introduced as a new member of the RAAS. 1 Its main biological function is converting Ang II into Ang (1–7), whose functional effects (vasodilation, anti-proliferation) are opposed to those of Ang II. 2
Animal models have shown that ACE2 is protective in heart failure. Recent studies in animal models have demonstrated that ACE2 overexpression attenuates cardiac remodeling and that treatment with Ang (1–7) prevents cardiac fibrosis.3–5 Clinical research findings were inconsistent. Some research reports that ACE2 gene was up-regulated in severe heart failure patients.6,7 but Batlle et al. reported there was no change in ACE2 expression in severe heart failure patients. 8 Because of the need for invasive myocardium sampling, little is known about the role of ACE/ACE2 balance in the development of human heart failure. We hypothesized that the endogenous regulatory arm of the renin-angiotensin-aldosterone system (RAAS) would be increased in patients with heart failure, that is, the balance between the counteracting enzymes ACE and ACE2 may change. In the present study, for the first time, we aim to determine the balance between ACE and ACE2 in patients with chronic heart failure (CHF).
Materials and methods
Subjects
In our study, we investigated 78 patients (47 men, 31 women; mean age, 59 years; age range, 41–73 years) with chronic heart failure (CHF) who were admitted to our hospital for myocardial biopsy. We evaluated the patients’ cardiac function according to the New York Heart Association (NYHA) classification. Patients with acute and chronic infection, diabetes, autoimmune diseases, malignant tumor, serious liver or kidney dysfunction or acute myocardial infarction (MI) were excluded from the study. Written informed consent was obtained from all patients and the study protocol was approved by our hospital’s institutional review board.
All clinical data were obtained by the same investigator. Patients with CHF received maximal medical therapy according to the treatment guideline recommendations for CHF. Almost all of the patients with CHF received an ACE inhibitor or angiotensin (Ang) receptor blocker at the highest tolerated doses (Table 1). One week before the operation, color Doppler echocardiography was performed in all patients to measure the left ventricular end diastolic diameter (LVED) and left ventricular ejection fraction (LVEF). According to NYHA classification, the patients were divided into the mild CHF group (NYHA II) or the moderate to severe CHF group (NYHA III–IV). The 13 subjects in the normal control group were organ donors who died in accidents. The differences in age and gender were not significant between patients and controls.
Patients’ general clinical and medical therapy data.

CHF: chronic heart failure; LVDd: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction; BNP: B-type natriuretic peptide; ACEI: angiotensin converting enzyme inhibitor; ARB: angiotensin receptor blocker.
Myocardial tissue collection
A small amount of left ventricular (LV) muscle tissue was collected from patients with dilated cardiomyopathy or ischemic cardiomyopathy. The control muscle tissue was collected 2 h after the accidental death of the donors. The samples were aliquoted immediately and frozen in liquid nitrogen for real-time polymerase chain reaction (PCR) and protein detection.
ACE and ACE2 mRNA expression by real-time reverse transcription (RT)-PCR analysis
We performed real-time RT-PCR analysis to assay ACE and ACE2 mRNA levels in left ventricles. Briefly, total RNA was extracted from the LV samples using Trizol reagent. Template cDNA was prepared using reverse transcriptase. The primers were synthesized by Race Parkson (Roche, Shanghai, China). The following primers were used to amplify the ACE: sense 5′-CCGATCTGGCAGAACTTC-3′ and antisense 5′-GTGTTCCAGATCGTCCTC-3′. The amplified product was 408 bp long. The following primers were used to amplify the ACE2: sense 5′-CATTGGAGCAAGTGTTGGATCTT-3′ and antisense 5′-GAGCTAATGCATGCCAT TCTCA-3′. The amplified product was 108 bp long. The primers for amplifying GAPDH were 5′-CGGATTTGGTCGTATTGGG-3′ and antisense 5′-TCTCGCTCCTGGAAGATGG-3′. Each sample was run in duplicate with an initial 10-min period at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The specificity and identity of the PCR products were validated by melting curve analysis.
Detection of ACE and ACE2 protein expression by western blotting analysis
The tissues were lysed with RIPA buffer and the protein concentration was measured using the Bradford method. A total of 250 μg of protein was separated on an 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel and transferred to a nitrocellulose membrane. The membrane was blocked using Tris-buffered saline containing 3% bovine serum albumin. Goat antibodies specifically against ACE or ACE2 (Santa Cruz; CA, USA) used at a dilution of 1:250 were incubated with the membranes for 1 h at room temperature. Secondary antibody (mouse anti-goat conjugated to horseradish peroxidase (Zhongshan Jinqiao, Beijing, China)) was incubated with the membranes for 1 h at room temperature. Proteins revealed by western blotting were visualized by chemiluminescence. After scanning, the optical density was analyzed using Quantity One software (Bio-Rad).
Statistical analysis
SPSS 13.0 was used to perform the statistical analyses. Data are presented as mean (standard deviation). Normal distribution was analyzed with Kolmogorov-Smirnov test. Levene’s test was used to test for equality of variances. One-way analysis of variance (LSD test) was employed to compare the differences between three groups. Values of P<0.05 were considered statistically significant.
Results
General clinical data of patients with CHF
The B-type natriuretic peptide and LVED values increased and the LVEF value decreased as the cardiac function decreased according to the NYHA classification system (Table 1).
The mRNA expression of ACE and ACE2
The mRNA expressions of ACE and ACE2 in the patients with CHF were significantly higher than those in normal controls (P<0.01, P<0.01,respectively). The mRNA expressions of ACE and ACE2 were significantly higher in patients with moderate to severe CHF than in those with mild CHF (P<0.01, P<0.01, respectively). The ratios of ACE/ACE2 mRNA decreased in patients with mild CHF but increased in patients with moderate to severe CHF (Table 2; Figure 1).
ACE and ACE2 mRNA and protein expression in patients with CHF.
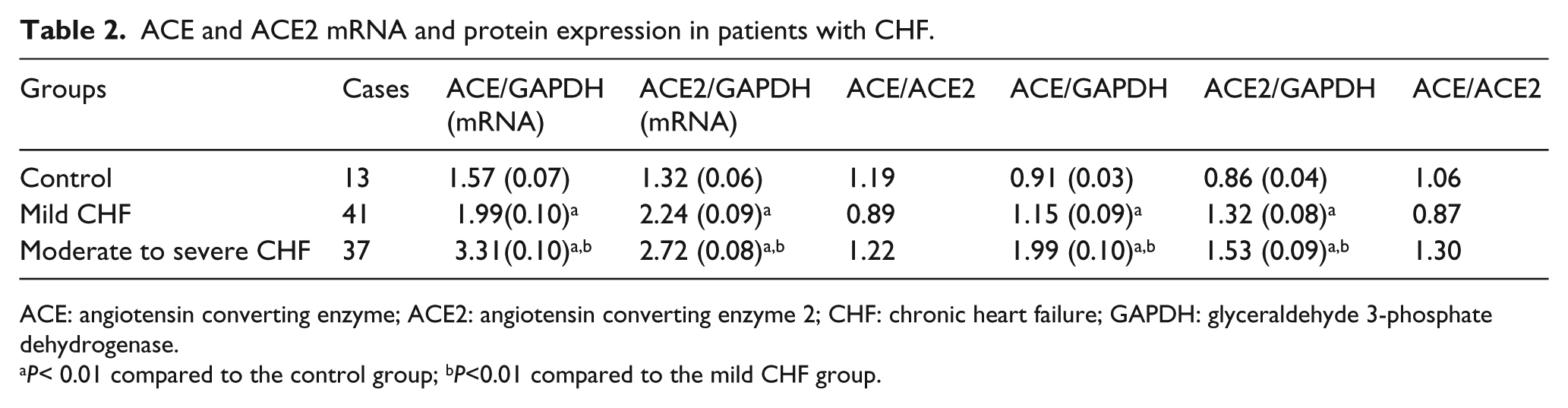
ACE: angiotensin converting enzyme; ACE2: angiotensin converting enzyme 2; CHF: chronic heart failure; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
P< 0.01 compared to the control group; bP<0.01 compared to the mild CHF group.

ACE and ACE2 mRNA expressions in the myocardial tissues of patients with CHF. The mRNA expressions of ACE and ACE2 in patients with CHF were significantly higher than those in normal controls (P<0.01). The mRNA expressions of ACE and ACE2 were significantly higher in patients with moderate to severe CHF than in patients with mild CHF (P<0.01). The ACE/ACE2 mRNA ratio decreased in patients with mild CHF but increased in patients with moderate to severe CHF. aP<0.01 compared to the control group; bP<0.01 compared to the mild CHF group.
ACE and ACE2 protein expression detected by western blotting
The protein expressions of ACE and ACE2 in patients with CHF were significantly higher than those in the normal controls (P<0.01, P<0.01, respectively). The protein expressions of ACE and ACE2 were significantly higher in patients with moderate to severe CHF than in patients with mild CHF (P<0.01, P<0.01, respectively). The ACE/ACE2 protein ratio decreased in patients with mild CHF but increased in patients with moderate to severe CHF (Table 2, Figure 2).

Detection of ACE and ACE2 protein expressions by western blotting.
Discussion
ACE2 is a newly discovered homolog of ACE that is highly expressed in the human heart. Its major biological function is to degrade Ang II to generate Ang (1–7).1,2 Ang (1–7) has vasodilatory, anti-proliferation, anti-inflammatory, and protective effects against heart failure.3,9–12 It was speculated that ACE2 might evolve as a natural counter-regulatory mechanism to inhibit Ang II overactivation, and ACE2/Ang (1–7) is a cardioprotective arm of the RAAS in CHF.4,5 We speculate that ACE and ACE2 are activated in severe heart failure, but in the presence of an ACE/ACE2 imbalance, increased ACE2 is unable to overcompensate Ang II, leading to further heart failure deterioration.
Since the discovery of ACE2, mounting evidence suggests that it plays an important role in regulating the physiological functions of the heart. Mice that are deficient in ACE2 have increased Ang II levels and a severe cardiac contractility defect. 5 In some studies using the aortic constriction model, ACE2 null mice developed enhanced susceptibility to biomechanical stress, increased pathological hypertrophy and worsening systolic performance. Myocardial Ang II levels were increased, whereas Ang 1–7 levels were decreased. AT(1) receptor blockade and Ang 1–7 decrease resulted in marked recovery of systolic dysfunction.13–15 Oudit et al showed that ACE2(-/y) mutant mice develop a progressive age-dependent dilated cardiomyopathy with increased oxidative stress, neutrophilic infiltration, inflammatory cytokines, and pathological hypertrophy. 16 Moreover, Song et al reported that ACE2 deficiency exacerbated Ang II-mediated myocardial injury. 17 ACE2 inhibition or loss has also been shown to facilitate adverse post-MI ventricular remodeling.18,19
These observations suggest that ACE2 could play a vital protective role in retarding the development of heart failure. For instance, some studies showed that ACE2 overexpression attenuated LV fibrosis, improved LV remodeling and systolic function, and inhibited expression of ACE, Ang II, and collagen I in a rat model of MI.20,21 Grobe et al delivered an ACE2 gene to cultured cardiac fibroblasts after acute hypoxic exposure, resulting in significant attenuation of hypoxia/re-oxygenation–induced collagen production by the fibroblasts. 22 In addition, Zhong et al. reported that treatment of Ang II-infused wild-type mice with recombinant human ACE2 blunted hypertrophy marker response and expression and reduced Ang II-induced superoxide production. 23
Despite many elegant animal studies, the role of ACE2 in the development of human heart failure is not well understood, and the need for invasive cardiac tissue sampling has limited clinical studies. Ohtsuki et al. and Goulter et al. reported that the ACE2 gene was up-regulated in the left ventricular myocardium of patients with severe heart failure and was associated with the degree of left ventricular dilatation.6,7 Moreover, Burrell et al. showed that MI results in increased ACE2 expression in both rats and humans. 24 For instance, after intensive medical therapy, a 50% increase in baseline serum ACE2 levels predicted a significant reduction in the risk of death, cardiac transplantation, or re-hospitalization. 25 Increased plasma ACE2 activities were strongly correlated with worsening LVEF fraction and increased B-type natriuretic peptide levels. ACE2 activity has no direct relationship with markers of systemic inflammation, and increased ACE2 activity is an independent predictor of the combined endpoints of heart failure hospitalization, cardiac transplant, and death.26–28
In agreement with previous reports, our study showed that ACE2 was upregulated in the left ventricular myocardium of patients with CHF was associated with the degree of left ventricular end-diastolic diameter and LVEF. However, the exact mechanisms of this phenomenon are not fully understood, suggesting that ACE2 expression may be activated as an adaptive compensatory mechanism to prevent myocardium remodeling. This finding sits well with the hypothesis that ACE2 may counterbalance the effects of RAAS stimulation.
This study was the first to find that the ACE/ACE2 ratio decreased in patients with mild CHF, which may be a compensatory response. Increased ACE2 expression could promote Ang II degradation and result in increased Ang (1–7) production to protect the failing heart. However, the ACE/ACE2 ratio increased in patients with moderate to severe CHF, and the ACE expression was relatively higher than the ACE2 expression, which resulted in more myocardial Ang II and expedited the heart failure. These associations were independent of other disease states and medication use, which suggests that the changing enzyme balance worsened the myocardial Ang II/Ang (1–7) balance, accelerating the progression of cardiac dysfunction. Due to the design of the experiemnet, our study hasn’t tested the levels for Ang II and Ang(1–7), which is the main limitation of the study. As the next step of our study, we will look into the level of the active factors including Ang II and Ang(1–7) among heart failure patients, so as to deeply reveal the role of RAAS in heart failure.
Since ACE2 can degrade Ang II and produce Ang (1–7), its activity is similar to those of the clinically widely used ACE inhibitors. This strengthens the hypothesis that ACE2 may be a relevant future target for the treatment of heart failure. Our data support further investigations to better understand the clinical utility and significance of targeting ACE2 pathways for therapeutic interventions.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.