
Abstract
Introduction:
The relationship between intestinal inflammation and circulating components of the renin-angiotensin system (RAS) is poorly understood.
Materials and methods:
Demographic and clinical data were obtained from healthy controls and patients with inflammatory bowel disease (IBD). Plasma concentrations of the classical RAS components (angiotensin-converting enzyme (ACE) and angiotensin II (Ang II)) and alternative RAS components (ACE2 and angiotensin (1-7) (Ang (1-7))) were analysed by radioimmuno- and enzymatic assays. Systemic inflammation was assessed using serum C-reactive protein (CRP), white cell count, platelet count and albumin, and intestinal inflammation by faecal calprotectin.
Results:
Nineteen healthy controls (11 female; mean age 38 years, range 23–68), 19 patients with Crohn’s disease (11 female; aged 45 years, range 23–76) and 15 patients with ulcerative colitis (6 female; aged 42 years, 26–64) were studied. Circulating classical RAS component levels were similar across the three groups, whereas ACE2 activity and Ang (1-7) concentrations were higher in patients with IBD compared to controls (ACE2: 21.5 vs 13.3 pmol/ml/min, p<0.05; Ang (1-7): 22.8 vs 14.1 pg/ml, p<0.001). Ang (1-7) correlated weakly with platelet and white cell counts, but not calprotectin or CRP, in patients with IBD.
Conclusions:
Circulating components of the alternative RAS are increased in patients with IBD.
Keywords
Introduction
Inflammatory bowel disease (IBD), comprising Crohn’s disease (CD) and ulcerative colitis (UC), are debilitating conditions affecting up to 0.5% of the population in Western countries and an increasing number in the developing world. 2 A significant proportion of patients fail to achieve remission with currently available therapies and have uncontrolled symptoms, penetrating or fibrostenosing complications, or experience adverse effects. This results in poor quality of life, requirement for surgery and has a significant health economic impact. 3 There is hence a strong impetus for ongoing investigation of disease pathogenesis to identify new therapeutic targets.
The renin-angiotensin system (RAS) is a hormonal system with a central role in cardiovascular and renal physiology. Angiotensin II (Ang II), the key effector peptide of this system, mediates vasoconstriction and sodium and water retention both directly and indirectly by stimulating aldosterone secretion. Ang II is implicated in the response to inflammation and regulation of tissue repair and fibrosis in many organs.4,5
Previously, the view of the RAS comprised a linear pathway involving the conversion of angiotensinogen to Ang II via a two-step process facilitated by renin and angiotensin-converting enzyme (ACE). Contemporary understanding suggests greater complexity (Supplementary Material, Figure 1). A homologue of ACE (a dipeptidase), known as ACE2 (a monopeptidase), was described over a decade ago. ACE2 has distinct enzyme activity and substrate affinity, and cleaves a single amino acid from Ang II to produce Ang (1-7).6,7 Ang (1-7) exerts its effects primarily via the mas receptor. Together, these components (ACE2/Ang (1-7)/mas) comprise the ‘alternative RAS’ pathway 8 distinguishing them from the ACE-Ang II-Angiotensin type 1 receptor (AT1R) pathway which has become known as the ‘classical RAS’. Ang (1-7) is demonstrated to have anti-inflammatory and anti-fibrotic properties that oppose the actions of Ang II.9–11 The classical and alternative RAS may represent complementary pathways, the balance of which determines whether healing or ongoing inflammation and tissue injury occur. 8 Though previously considered an endocrine system, recent evidence demonstrates the RAS has paracrine and autocrine functions with components constitutionally expressed in numerous organs including the heart, kidney, brain, pancreas and liver12–14 with upregulation in chronic inflammation. Drugs that modulate RAS enzymes and receptors have been therapeutically applied in diabetes, cardiac, renal and liver disease.
Little is known regarding the role of the RAS in IBD. Many components of the classical RAS have been identified throughout the gastrointestinal tract, with functional influence on a range of processes including secretion and absorption of fluid, electrolytes, glucose and peptides, gastrointestinal motility and mesenteric blood flow. 1 Concentrations of ACE and ACE2 mRNA in the terminal ileum and colon are higher than in most other human tissues, 15 with potential implications for regulation of inflammation and fibrosis in IBD. Within the human colon, the AT1R and AT2 receptor (AT2R) have been localised in vessel walls, surface epithelium, crypt bases and myofibroblasts and macrophages within the lamina propria. 16
Few studies have examined expression of the RAS in patients with IBD to elicit its contribution to disease pathogenesis. Perturbed circulating ACE in sarcoidosis and other granulomatous conditions led to its study in CD, but the findings were largely conflicting; with some finding reduced levels17–20 whilst others noted no difference compared with healthy controls.21–23 Neither circulating Ang II nor the components of the alternative RAS have previously been studied in IBD. Elevated plasma Ang (1-7) levels are noted in patients with hepatic inflammation secondary to hepatitis C, 24 children with hypertension, chronic kidney disease and pregnant women,25,26 in whom it may mediate anti-inflammatory effects and compensatory vasodilatation. 27
The objectives of this study, therefore, were to comprehensively characterise the circulating components of the classical and alternative RAS in patients with CD and UC compared to healthy controls, and to examine the relationship of the RAS with markers of disease activity.
Methods
Subjects
Patients with CD and UC were recruited via a letter of invitation given to consecutive patients attending the Eastern Health IBD Clinics. Healthy controls were recruited via invitation to staff members by email within the Department of Gastroenterology and Hepatology at Eastern Health and Eastern Health Clinical School. Subjects taking an ACE inhibitor (ACEi) or angiotensin receptor blocker (ARB) were excluded. Written informed consent was obtained from all participants. The protocol for this study was approved by the Eastern Health Department of Research and Ethics. The research materials related to this paper are stored securely within Eastern Health Clinical School, Monash University.
Protocol
Information regarding gastrointestinal symptoms, co-morbid illnesses, medications, and lifestyle factors including physical activity was collected. Patients with IBD were characterised according to the Montreal Classification of IBD. 28 Symptomatic disease activity was evaluated by the Harvey Bradshaw Index (HBI) and Simple Clinical Colitis Activity Index (SCCAI) for patients with CD and UC, respectively. Cardiovascular indices (blood pressure, heart rate) and anthropological features (height, weight and waist circumference) were recorded. Peripheral blood was collected via venepuncture from the antecubital fossa with the patient in the sitting position after being upright (sitting, standing, walking) for at least two hours. Plasma collected in 4 ml lithium heparin tubes was stored at –80oC prior to analysis for ACE2. For angiotensin peptides, plasma was collected in 4 ml Na2 ethylenediaminetetraacetic acid (EDTA) tubes, and enzyme inhibitor mix consisting of 85 µl Na2 EDTA 50 µM, aprotinin 21000 U/ml, N-ethylamide 0.2 M, leupeptin 10.5 µM and pepstatin-A 1.5 µM was added. These were stored at –20oC until analysis. All other analyses were performed on fresh serum and plasma as appropriate. A faecal sample obtained within the previous 24 h was stored at –20oC.
Analytical assays
Blood samples were analysed for laboratory indices including electrolytes, renal and liver function, and markers of systemic inflammation (CRP, white cell count, platelet count and albumin) via routine laboratory techniques. Serum ACE was measured using Infinity ACE Liquid Stable Reagent (ThermoScientific, Waltham, Massachusetts, USA) by optical density using manufacturer’s instructions. Values were expressed in U/l, with a reference range 8–52 U/l. Plasma renin activity was measured by the Gammacoat Plasma Renin Activity Radioimmunosassay Kit (Diasorin Inc., Stillwater, Minnesota, USA), as per manufacturer’s instructions. Values were expressed in µg/l/h, with a reference range 1.31–3.95 µg/l/h. Serum aldosterone was calculated using the Coat-a-count Aldosterone Solid Phase Radioimmunoassay (Siemens, Los Angeles, California, USA) as per manufacturer’s instructions, with values expressed in pmol/l, and reference range 110–860 pmol/l.
Plasma ACE2 activity was measured using a quenched fluorescent substrate-based assay (QFS: (7-methoxy-coumarin-4-yl) acetyl-Ala-Pro-Lys-(2,4-dinitrophenyl)-OH, Auspep, Parkville, Victoria, Australia), as described previously.29,30 Briefly, 250 µl of plasma underwent an anion exchange process using low-ionic-strength buffer (20 mmol/l Tris-HCl, pH 6.5) and ANX sepharose 4 Fast-Flowresin (agarose beads, GE Healthcare Biosciences Australia). Following the extraction process, 100 µl of eluate were incubated with 20 µl 50 mM QFS. Samples were analysed in duplicate with and without 100 µM EDTA as ACE2 inhibitor in a final volume of 200 µl per well. Fluorescence generated from the cleavage of QFS by ACE2 was continuously recorded by a fluorescent plate reader at excitation and emission wavelengths of 320 nm and 405 nm respectively (FluoStar Optima plate reader, BMG Labtech, Offenburg, Germany). The rate of substrate cleavage was determined by comparison to a standard curve of the free fluorophore, 4-amino-methoxycoumarin (MCA; Sigma, Missouri, USA) and expressed as pmol of substrate cleaved/ml of plasma/min. The intra-assay coefficient of variation was 5.6% (n=20) and the inter-assay coefficient of variation was 11.8% (n=14).
Plasma Ang II and Ang (1-7) were measured using a direct radioimmunoassay, performed on all samples in duplicate (ProSearch International Australia Pty Ltd, Malvern, Victoria, Australia) as described previously. 31 Results were expressed in pg/ml.
Intestinal inflammation was assessed by faecal calprotectin using Quantum Blue Calprotectin quantitative lateral flow assay (Bühlmann Laboratories, Schonenbüch, Switzerland) within one week of collection, as per manufacturer’s instructions. Faecal calprotectin was quantified by Buhlmann Quantum Blue Reader, with values expressed in µg/g faeces. Values were quantified within the range of 30–300 µg/g, with values below this range expressed as <30 µg/g. Specimens with faecal calprotectin >300 µg/g were quantified using Buhlmann Quantum Blue High Range Calprotectin assay following dilution to 1:300.
Definition of active disease
No single clinical or biomarker-based index has been shown to accurately distinguish between disease remission and active disease in patients with IBD. In the present study, disease activity in patients with IBD was assessed via multiple parameters, including clinical indices (HBI, SCCAI), serological parameters (CRP, white cell count, platelet count and albumin), and faecal calprotectin. In the absence of endoscopic assessment, active disease was objectively defined by a faecal calprotectin ≥100 µg/g and/or a CRP ≥5 mg/l, and remission hence defined as faecal calprotectin <100 µg/g and a CRP <5 mg/l. Faecal calprotectin has been shown to correlate with disease activity in patients with IBD, but the sensitivity and specificity varies depending on the cut-off used. A cut-off of 100 µg/g has been shown to be most discriminatory in studies to date,32–35 though other levels of calprotectin have been assessed.36–39
Statistical analyses
Statistical analyses were performed using SPSS v20 (IBM Corporation, 2011) and GraphPad Prism v5.04 (Graphpad software, 2010). Analysis of variance (ANOVA) and unpaired t-tests (two-sided) were used for comparison of means between groups, with potential confounding variables adjusted for using multivariate analysis of covariance (MANOVA). The relationship between variables was assessed by bivariate and partial correlation using Pearson’s coefficient for parametric and Spearman’s coefficient for non-parametric variables as appropriate. Values for faecal calprotectin were normalised by log transformation. Receiver-operator characteristic (ROC) curves for ability to discriminate between patients with IBD and healthy controls were generated for faecal calprotectin, CRP, Ang (1-7) and ACE2. A p-value of 0.05 or less was considered statistically significant.
Results
Subjects
Nineteen healthy controls, 19 patients with CD and 15 with UC were recruited. As shown in Table 1, no significant demographic differences were noted across the three groups. However, patients with IBD had greater body mass index (BMI) and waist circumference, and a higher resting heart rate than healthy controls. None of the participants were taking other medications that may influence circulating concentrations of RAS components, such as ß-receptor antagonists, calcium channel antagonists or diuretics.
Baseline characteristics and laboratory markers.

ANOVA: analysis of variance; BMI: body mass index; BP: blood pressure; CI: confidence interval; CKD: chronic kidney disease; DM: diabetes mellitus; NCS: neurocardiogenic syncope; DM: diabetes mellitus; NS: not significant.
Characteristics of patients with IBD are shown in Table 2. Most patients with CD had ileocolonic disease (58%), and had either stricturing (Montreal Classification B2, 21%) or penetrating disease (Montreal Classification B3, 58%), with perianal disease present in 58% of patients. Active disease defined by a faecal calprotectin of ≥100 µg/g and/or a C-reactive protein of ≥ 5 mg/l was present in 47% of patients with CD, and 84% were on treatment with immunomodulators and / or anti-tumour necrosis factor (TNFα) medications. Fourteen patients (42%) had previously required intestinal resection. Amongst patients with UC, 13% of patients were defined as being in clinical remission, 73% as having mild disease and a minority (14%) as having moderate or severe disease according to the Montreal Classification of severity. However, objective biomarker (faecal calprotectin ≥100 µg/g and/or a C-reactive protein of ≥5 mg/l) evidence of active disease was present in only 60% of patients. No patients with UC had previously had a colectomy.
Characteristics of patients with inflammatory bowel disease (IBD).

5-ASAs: 5-aminosalicylates; 6-MP: 6-mercaptopurine; ADA: adalimumab; GI: gastro-intestinal; IFX: infliximab.
Of the markers of inflammation, the total white cell count was higher in patients with CD than in the other groups, and serum albumin was lower and faecal calprotectin higher in patients with IBD compared to healthy controls (Table 3).
Routine laboratory indices in the patient groups and healthy controls.
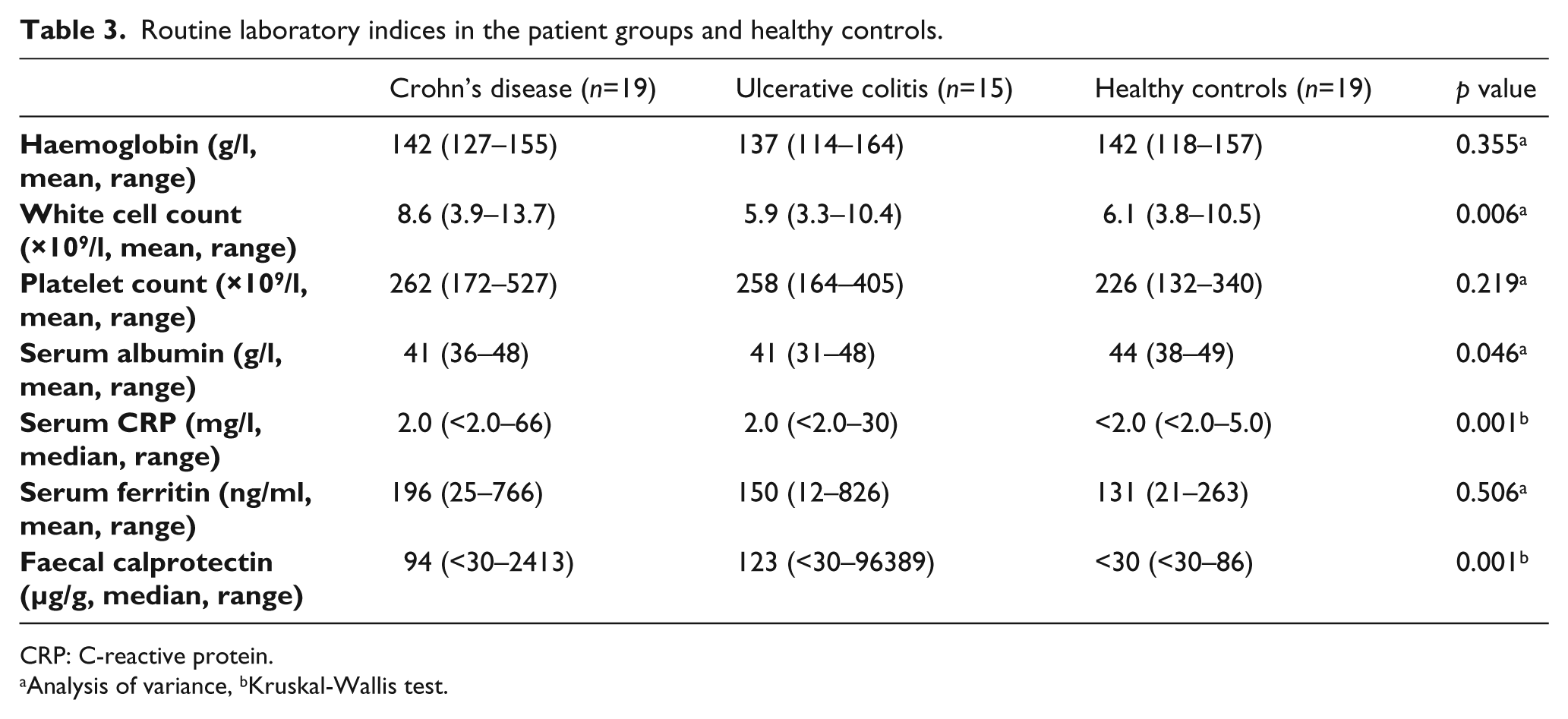
CRP: C-reactive protein.
Analysis of variance, bKruskal-Wallis test.
Comparison of circulating RAS components in patients with IBD and healthy controls
Circulating concentrations of RAS components in patients with IBD and healthy controls are shown in Figure 1. Compared to those in healthy controls, aldosterone levels were lower in patients with UC, and the aldosterone:renin ratios significantly lower in all patients with IBD; however, these differences were not significant after adjustment for corticosteroid use, which may have mild mineralocorticoid activity. There were no disease differences in circulating classical RAS components, ACE and Ang II.
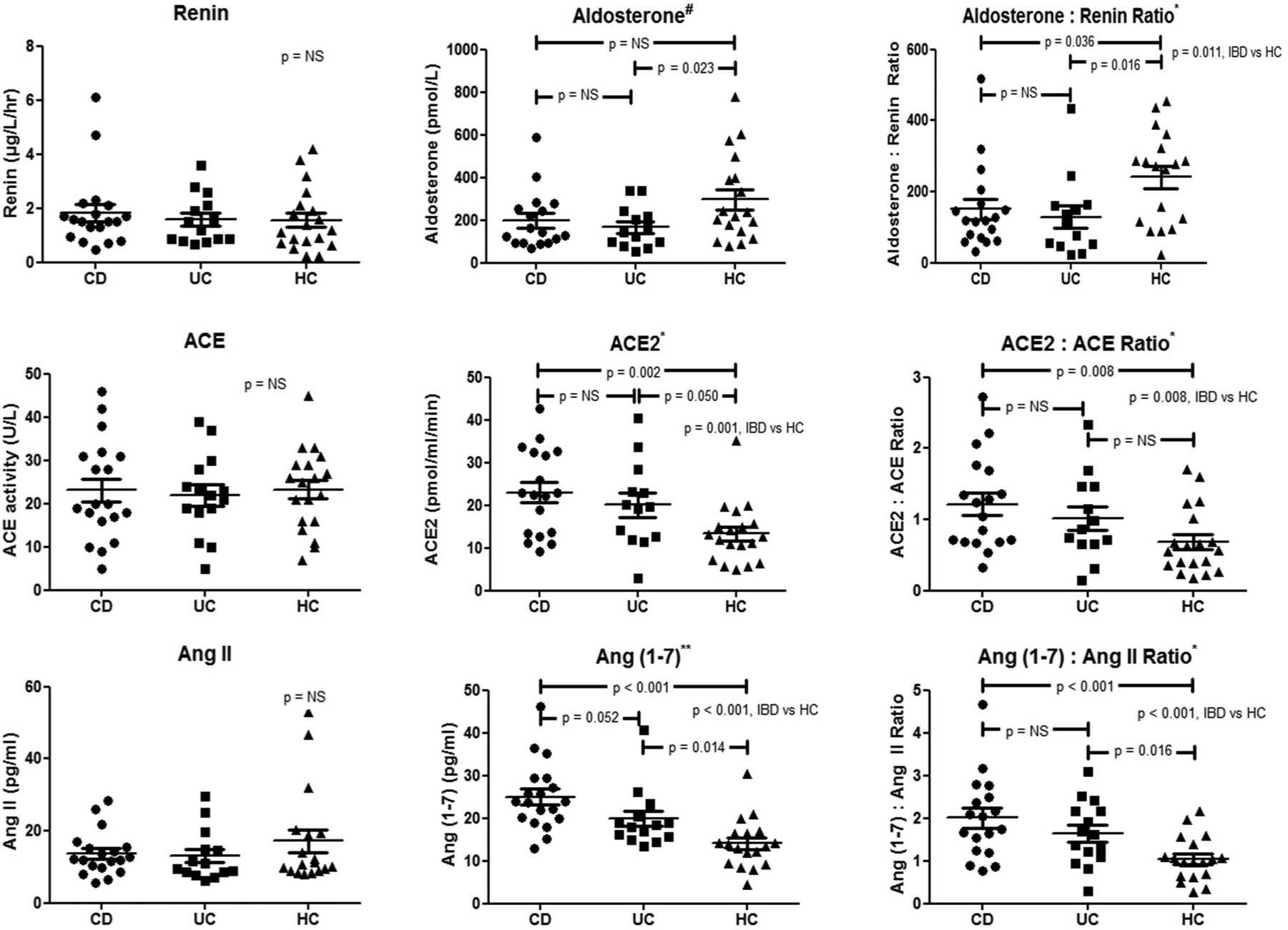
Circulating renin-angiotensin system (RAS) components in patients with inflammatory bowel disease (IBD). Analysis of variance (ANOVA): #p<0.10, *p<0.05, **p<0.001. ACE: angiotensin-converting enzyme; Ang: angiotensin; CD: Crohn’s disease; HC: healthy control; NS: not significant; UC: ulcerative colitis
Notably, the alternative RAS components, ACE2 (p=0.001) and Ang (1-7) (p<0.001), and the ratios of ACE2:ACE (p=0.008), and Ang (1-7):Ang II (p<0.001) were higher in patients with IBD than in healthy controls (Figure 2). No significant effect of concomitant steroid, immunosuppressive or biological therapy was evident (data not shown). Both subgroups of patients with active IBD (as defined by faecal calprotectin ≥100 µg/g and/or C-reactive protein ≥5 mg/l, n=18) and quiescent IBD (faecal calprotectin <100 µg/g and C-reactive protein <5 mg/l, n=16) had higher levels of Ang (1-7) and ACE2 than healthy controls (Table 4).

Correlation of angiotensin 1-7 (Ang (1-7)) with markers of disease activity in (a) patients with inflammatory bowel disease (IBD), (b) Crohn’s disease (CD) and (c) ulcerative colitis (UC). CRP: C-reactive protein; NS: not significant; SCCAI: Simple Clinical Colitis Activity Index; WCC: white cell count.
Comparison of angiotensin (1–7) (Ang (1–7)) and angiotensin-converting enzyme 2 (ACE2) levels between subgroups of patients with inflammatory bowel disease (IBD) with active disease (faecal calprotectin ≥100 µg/g and/or C-reactive protein≥5 mg/l) and quiescent disease (faecal calprotectin<100 µg/g and C-reactive protein<5 mg/l) with healthy controls.

CI: confidence interval.
Independent samples t-test.
On ROC analysis, Ang (1-7) (area under curve (AUC) 0.852) showed similar discriminant ability between patients with IBD and healthy controls to faecal calprotectin (AUC 0.846), and was superior to CRP (AUC 0.625). Plasma ACE2 levels showed weak-moderate discriminant ability (AUC 0.692).
Across all participants, there were significant correlations between ACE2 and age (Pearson r=0.30, p=0.032), body mass index (r=0.42, p=0.003), waist circumference (r=0.40, p=0.004), and systolic blood pressure (r=0.48, p=0.001). Ang (1-7) was noted to correlate with heart rate (r=0.32, p=0.023). Hence, to adjust for the effect of these potential confounders, a model for MANOVA was developed using ACE2 and Ang (1-7) as dependent variables, diagnosis as independent variable and co-variates comprising age, body mass index, systolic blood pressure and heart rate. The differences between groups remained significant (Pillai’s trace=0.304, p=0.005). Considering Ang (1-7) and ACE2 separately on tests for between-subjects effects in this model, only Ang (1-7) remained statistically significant (p<0.001).
Correlation of circulating RAS components with markers of disease activity
None of ACE, Ang II or plasma ACE2 activity showed statistically significant correlation with any markers of disease activity in patients with IBD. However, concentrations of Ang (1-7) significantly correlated, albeit weakly, with platelet and white cell counts, but not with faecal calprotectin or CRP (Figure 2). In patients with UC, Ang (1-7) correlated with white cell and platelet counts and SCCAI, but not CRP, faecal calprotectin or albumin.
Plasma renin activity and aldosterone:renin ratio did not significantly correlate with any indices of disease activity. A positive correlation between aldosterone and white cell count in patients with UC was noted, but was no longer significant after excluding patients concurrently treated with corticosteroids (n=3), which may result in both leukocytosis and mild mineralocorticoid activity.
Given the established role of the RAS in inflammatory and fibrotic processes in other organ systems, the subgroup of patients with CD with a stenosing and fistulising phenotype (n=16) was separately analysed. None of the RAS components studied reached statistically significant correlations with the indices of disease activity in this subgroup.
Discussion
The role of the RAS in health and disease is well defined in a number of organs, but is poorly characterised in the gastrointestinal tract. The current pilot study addressed the possible involvement of the RAS in IBD pathogenesis by examining the circulating concentrations of its key elements. The study provides the first evidence that the alternative RAS components, ACE2 and Ang (1-7), are upregulated in patients with both CD and UC, but have no clear correlation with degree of disease activity.
These findings have potentially intriguing implications regarding understanding of the mechanism of inflammation in IBD, and suggest possible new therapeutic targets. First, upregulation of Ang (1-7) and ACE2 may imply a compensatory response to intestinal inflammation given the described anti-inflammatory functions of Ang (1-7) in various organs. The precise source of circulating ACE2 or Ang (1-7) in this setting is uncertain, and cannot be determined from the current study. Circulating ACE2 concentrations have recently also been noted to be elevated in patients with type 1 diabetes, and correlated with measures of arterial stiffness, systolic blood pressure, duration of disease and inversely with eGFR.40,41 It has been postulated that the metallosecretase, ADAM 17, may cleave vascular membrane ACE2 at its ectodomain and hence promote its release into the circulation. 42 Therefore, systemic ACE2 upregulation may reflect underlying vascular or endothelial stress, a common feature in many inflammatory conditions, including IBD. 43 Mean resting heart rate was noted to be higher in patients with both quiescent (79 beats per min, 95% confidence interval (CI) 74–85 bpm; p<0.001) and active (77 bpm, 95% CI 71–83 bpm; p=0.007) than healthy controls (66 bpm, 95% CI 62–70 bpm). Furthermore, no correlation between ACE2 and heart rate (p=0.14, p=0.433) was noted amongst patients with IBD, despite an association between Ang (1-7) and heart rate (r=0.32, p=0.023). Any potential relationship between cardiac stress and ACE2 is therefore unclear from the data in this study. Though it is accepted that active IBD may result in tachycardia, the reasons for a higher heart rate in patients with quiescent disease are less certain. Given the small cohort in this study, the differences may be confounded by a greater weight, reduced physical fitness or anxiety associated with the study visit amongst patients with IBD than healthy controls.
Of particular pertinence to IBD, ACE2 is present in the terminal ileum and colon in concentrations that are amongst the highest in the body. 15 As well as the vasculature, it is also found at the brush border of enterocytes. 44 Furthermore, Ang II has been noted to be elevated in rectosigmoid biopsies from patients with Crohn’s colitis compared with patients with UC or healthy controls, and significantly correlated with endoscopic grade of colitis. 45 Hence, co-localisation of ACE2 and Ang II demonstrates capacity to generate Ang (1-7) with compensatory counter-regulation of inflammation. Studies to localise and quantify ACE2 and Ang (1-7) in the intestinal mucosa, and correlate these with circulating levels, in patients with IBD, will help to shed further light on this issue.
As an alternative hypothesis, Ang (1-7) may itself be a mediator of inflammation under certain circumstances. Though most evidence indicates Ang (1-7) has anti-inflammatory and anti-fibrotic effects, recent studies suggest greater complexity in this system. In human mesangial cells and models of nephropathy, Ang (1-7) stimulates certain pro-inflammatory pathways.46,47 Clearly, functional studies of the effect of Ang II and Ang (1-7) and specific receptor antagonists in intestinal mucosal inflammatory models will be required to explicate this paradox as well as establish cellular mechanisms of effect.
Secondly, the potential role of the RAS in mucosal inflammation in IBD provides an exciting opportunity for therapeutic intervention. ACEi and ARBs have an established role as anti-hypertensive agents, and ameliorate cardiac and renal inflammation and fibrosis. These drugs have been shown to produce a number of beneficial anti-inflammatory effects in rodent models of intestinal inflammation.48–53 Though originally thought to exert their action predominantly via blockade of the effects of Ang II, recent evidence suggests some of the beneficial effects may be through upregulation of the alternative RAS with increased tissue Ang (1-7).54–56 Hence, if translated to human trials in IBD, these inexpensive, readily available medications may be suitable for treatment of inflammation and fibrosis in IBD.
Despite clear differences in the concentration of Ang (1-7) between patients with IBD and healthy controls, no unequivocal relationship between Ang (1-7) and markers of disease activity was demonstrated. There are two potential explanations for this finding. First, it may reflect the relatively small cohort analysed (type II error); this is supported by the finding of a significant correlation between Ang (1-7) and platelet and white cell counts. Secondly, and perhaps more fascinating, upregulation of the alternative RAS may occur at an early stage in pathogenesis of IBD, and is hence independent of the degree of inflammation; or persists following resolution of inflammation due to altered intestinal vascularity. The latter theory is supported by the fact that, as well as patients with active disease, patients with quiescent disease, as defined by faecal calprotectin < 100 µg/g and CRP <5 mg/l (n=16), had higher levels of Ang (1-7) (p=0.001) and ACE2 (p=0.003) than healthy controls. As discussed above, the source of circulating ACE2 and Ang (1-7) is important to investigate in this regard, as is mucosal localisation and quantification of these components in quiescent and active IBD.
A possibility that requires further exploration is the role of circulating concentrations of Ang (1-7) and ACE2 as biomarkers to identify patients with IBD, with ROC analysis revealing AUC of 0.852 and 0.692 respectively. These results compare well with the currently accepted faecal calprotectin (AUC 0.846) and CRP (AUC 0.625). While the current study examined a small population, the disease activity did vary considerably and even patients with quiescent disease exhibited higher levels of Ang (1-7) and ACE2. Given potential confounders in this study, the accuracy of these tests will need to be assessed in much larger cohorts, including in patients with non-IBD related inflammatory diseases. Though relatively inexpensive, these assays require specific methodology–immediate mixing after collection with enzyme inhibitors in the case of Ang (1-7), and quenched fluorescent substrate assay for ACE2–that may limit widespread applicability.
Of potential importance, the association of circulating concentrations of ACE2 with age, BMI and waist circumference has not previously been described. Given that the healthy controls in this cohort had lower BMI, waist circumference and resting heart rate, adjustment for these potential confounders on multivariate analysis was performed. Though statistically significant differences in ACE2 no longer remained, the difference in circulating Ang (1-7) was maintained. Nonetheless, the relationship of alternative RAS components with markers of obesity, which is itself a pro-inflammatory state, warrants further investigation using a larger and adequately powered sample of a healthy population.
In conclusion, circulating components of the RAS are perturbed in patients with IBD with an upregulation of the alternative RAS enzyme, ACE2, and the peptide, Ang (1-7). Quantification of ACE2 and Ang (1-7) also has potential as biomarkers for intestinal inflammation. Further exploration of these components in larger cohorts and at the level of mucosa, with their localisation and evaluation of function, are warranted to establish cellular mechanistic pathways. Such studies may enhance our understanding of the pathogenesis of IBD and provide a new class of agents to target intestinal inflammation and fibrosis.
Footnotes
Acknowledgements
The authors would like to thank Catherine Smith for statistical assistance with this paper, Amy Clements for assistance with faecal calprotectin assays and David Casley for assistance with Ang II and Ang (1-7) radioimmunoassays. MG carried out intellectual planning, literature review, patient recruitment, collection of specimens and data, analysis of RAS components, analysis and writing of the article. LB participated in intellectual planning and critical appraisal of the article. EV assisted with analysis of samples for ACE2 and critical appraisal of the article. KG assisted with analysis of samples for ACE2, and review of the article. PA participated in intellectual planning and critical appraisal of the article. PG participated in intellectual planning and critical appraisal of the article. JL participated in intellectual planning and critical appraisal of the article. All authors read and approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.