
Abstract
Introduction:
The present study was designed to investigate the effect of ramipril (angiotensin-converting enzyme inhibitor) on the chronic constriction injury of sciatic nerve induced neuropathic pain in mice.
Methods:
The neuropathic pain was induced by four loose ligations of the right sciatic nerve in mice. The battery of behavioral tests, i.e. plantar, pin prick, tail flick, tail pinch, rota rod tests, were performed to assess the degree of thermal and mechanical hyperalgesia in ipsilateral paw and tail, and motor in-coordination activity respectively. In addition, the biochemical tests, i.e. total protein, thiobarbituric acid reactive substances and reduced glutathione, were also performed in sciatic nerve tissue samples.
Results:
The administration of ramipril (2 and 4 mg/kg, p.o.) significantly attenuated chronic constriction injury-induced rise in peripheral as well as central pain sensitivity (thermal and mechanical) along with impairment of motor in-coordination activity. Further, it also produces ameliorative effects on chronic constriction injury-induced rise in thiobarbituric acid reactive substances and decrease in glutathione levels when compared with a normal control group.
Conclusion:
It may be concluded that angiotensin-converting enzyme inhibitor may be a potential new target for the management of neuropathic pain.
Keywords
Introduction
Pain is a warning signal in our body which elicits during the abnormality of the physiological process,1,2 whereas neuropathic pain is not a symptom, but a disease itself. Generally, it occurs due to damage of the nervous system.3,4 The International Association for the Study of Pain (IASP) defined neuropathic pain as ‘pain initiated or caused by a primary lesion or dysfunction in the nervous system’.5,6 The damage of the nervous system produces pain sensations such as hyperalgesia, allodynia 7 and spontaneous pain. 8 Clinically, neuropathic pain is managed by various conventional medicines, i.e. anti-depressants (amitryptyline, nortriptyline, lofepramine, duloxetine and venlafaxine); anti-convulsants (gabapentin and pregabalin); opioids (morphine, oxycodone, fentanyl, buprenorphine and methadone) and topical agents (lidocaine, tramadol, codeine and dihydrocodiene). 9 However, long-term administration inevitably produces unwanted side effects, i.e. dizziness, blurred vision, palpitation, sedation, urinary retention and reduced appetite. 9
Recent studies have documented the chronic injury of the peripheral nerve, which produces activation of the sympathetic (efferent) nervous system10,11 followed by the enhanced release of renin, angiotensin and aldosterone via the renin-angiotensin-aldosterone (RAAS) pathway.12–14 Further, angiotensin plays a critical role in neuronal damage.15,16 The RAAS modulators have been reported to ameliorate the neuropathic pain symptoms in various experimental animal models, i.e. aliskiren, a direct renin inhibitor; 17 ZD-7155, an angiotensin I receptor antagonist; 18 EMA401 and EMA300, an angiotensin II receptor antagonist;19,20 and spironolactone, an aldosterone antagonist. 21
Angiotensin-converting enzyme (ACE) inhibitors enhance the bradykinin level and can cause the pain sensitivity via nociceptor (bradykinin receptor). Bradykinin has pleiotrophic actions, and potent physiological effects, i.e. decreased blood pressure, increased vascular permeability and the promotion of classical symptoms of inflammation such as vasodilation, hyperthermia, edema and pain (warning signal). In addition, it has beneficial effects, i.e. potent anti-thrombogenic, anti-proliferative and anti-fibrogenic effects. 22 Bradykinin is an endogenous nonapeptide which is catabolised by ACE. ACE inhibitors increase the level of bradykinin by reduction of catabolic action of BK(1-9). BK(1-9) produces the pain sensation by direct stimulation of nociceptors in the peripheral nervous system but this effect is of very short duration of action and its half-life is very short (15 seconds). 23 In addition, BK(1-9) is not only degraded by ACE but also by other enzymatic pathways, i.e. aminopeptidase P, carboxypeptidase, dipeptidyl peptidase IV, endothelin-converting enzyme, neprilysin, propyl oligopeptidase, and the neutral endopeptidase.23,24 Furthermore, bradykinin has also been reported to produce pro- and anti-nociceptive action in a dose dependent manner. 25 The lipophilic ACE inhibitor rapidly crosses the central nervous system, and its half-life in cerebrospinal fluid (CSF) is 0.5–0.75 h.26,27 In addition, ACE inhibitor, i.e. ramipril, has pleiotropic effects, which is not equally shared by other ACE inhibitors. Moreover, ramipril also increases the endothelium-dependent relaxation factor (i.e. nitric oxide) and vasodilatation via B2 receptors. 28 Ramipril has a potential role in cardiovascular outcomes along with neuroprotective action. 29
However, the RAAS modulator is known to produce potential side effects, such as angioedema, hypotension and acute pancreatitis by angiotensin receptor blocker, renin inhibitor and ACE inhibitors.30–32 ACE inhibitors and angiotensin receptor blockers both cause cough and angioedema which produces cardiovascular risk in human.33,34 Angiotensin II is more prone to causing vasoconstriction along with ischemic neuropathy by activation of the angiotensin receptor in the nervous system. 35 Angiotensin II formation is due to ACE activity. The reduction of angiotensin II may be beneficial in neurodegenerative disorders such as neuropathic pain, instead of angiotensin receptor blockers and renin inhibitors.17,35 However, the role of ACE inhibitors has not yet been explored in the chronic constriction injury of sciatic nerve induced neuropathic pain model in mice. Therefore, the present study is focused to evaluate ramipril (ARTERE®; ACE inhibitor) in the management of chronic constriction injury-induced neuropathic pain in mice.
Materials and method
Drugs and chemicals
Folin-Ciocalteu’s phenol reagent (Merck Limited, Mumbai), 5,5’-dithio-bis-(2-nitrobenzoic acid (DTNB), 1,1’,3,3’ tetramethoxy propane, reduced glutathione and bovine serum albumin (BSA) (Sisco Research Laboratories Pvt. Ltd. Mumbai) were procured for the present study. DTNB was dissolved in 1% w/v of sodium citrate. All the reagents used in the present study were of analytical grade.
Animals
Swiss mice weighing 25–30 g were employed in the present study. Animals were maintained with a standard laboratory diet (Markfed cotton seed processing plant, Gidderbaha, Mukatsar, Punjab, India) and water ad libtium. Further animals were exposed to the natural light and dark cycle. The experimental protocol was approved by the Institutional Animal Ethics Committee (IAEC) and care of the animals was taken as per the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environmental and Forest, Government of India (Reg. no. 1407/a/11/CPCSEA).
Induction of peripheral neuropathy by chronic constriction injury
Neuropathic pain was induced in mice by chronic constriction injury of the sciatic nerve as described by Ma and Eisenach. 36 Briefly, mice were deeply anesthetized with thiopental sodium (35 mg/kg, i.p.). The hair from the lower back in the thigh region was removed and the skin was sterilized with 0.5% w/v povidine solution. The skin and muscle layer of the lateral surface of the right thigh was incised and the sciatic nerve was exposed. The four loose ligatures (silk thread no. 4) were placed around the proximal portion of the sciatic nerve from the trifurcation part (i.e. near knee joint), and a distance of 1 mm was maintained between each ligature. The loose ligatures were applied until a short flick response of the ipsilateral hind paw. After completion of the chronic constriction injury (CCI) procedure, muscle and skin layer was immediately sutured with silk thread and topical antibiotic powder was applied at once. Nociceptive threshold was assessed at different time intervals: 0, 1, 3, 7 and 14 days.
Behavioral evaluation
Plantar test
Thermal (radiant) hyperalgesia was assessed as described by the method of Hargreaves et al. 37 Briefly, the right hind paw of the mouse was placed on the radiant heat source. The thermal sensitivity of the hind paws was observed as the degree of hind paw withdrawal latency from the radiant heat lamp source. Withdrawal of the hind limb was considered as a positive response. The cut-off time was maintained at 20 seconds.
Pin prick test
Mechanical hyperalgesia was assessed as described by the method of Erichsen and Blackburn-Munro. 38 A blunted needle was applied to the mid plantar surface of the right hind paw at an intensity sufficient to produce a reflex withdrawal response. The needle was applied six times per minute. Withdrawal of the hind limb was considered as positive response. The cut off stimuli was applied six times to avoid the tissue injury and wind-up phenomenon.
Tail flick test
Tail thermal (radiant) hyperalgesia was assessed as described by the method of D’Amour and Smith 39 with the slight modification of Hargreaves et al. 37 Briefly, the tail of the mouse was placed on the radiant heat source. The thermal sensitivity of the tail was observed as the degree of tail withdrawal latency from the radiant heat lamp source. Withdrawal of the tail was considered as positive response. The cut-off time was maintained at 15 seconds to avoid tail damage.
Tail pinch test
Mechanical stimulus induced pain sensitivity was assessed by the tail pinch test according to modified Heffner’s method as described by Takagi et al. 40 Briefly, mice were tested by pinching their tail base with a Hoffmann clamp adjusted to elicit a nociceptive response. The nociceptive response was indicated by the number of attempts at the dislodgment of the Heffner’s clamp. The pressure stimulus evoked nociceptive response was noted and a cut-off time of 1 minute was maintained to prevent tissue damage.
Rota rod test
The motor in-coordination was assessed as described by the method of Jones and Roberts 41 with the slight modification of Muthuraman et al. 42 Briefly, mice were placed on the rotating rod (25 rpm) after a training period (i.e. 5 min) before 30 minutes of motor in-coordination test. The motor in-coordination of mice was assessed as the degree of fall in time from the rotating rod. The cut off time was maintained at 5 minutes to avoid potential muscular damage.
Biochemical analysis
All of the groups of animals were sacrificed after 14 days by cervical dislocation and complete sciatic nerves were isolated immediately. All parts of nerves were used for biochemical estimations. The sciatic nerve was homogenated (10% w/v) with phosphate buffer (pH 7.4) and centrifuged at 3500 rpm for 10 min. The supernatant was used for the estimation of tissue total protein, thiobarbituric acid reactive substances (TBARS) and reduced glutathione (GSH) levels.
Estimation of total protein content
The tissue total protein content was estimated as described by the method of Lowry et al. 43 Briefly, 300 µl of supernatant was diluted up to 1 ml with distilled water in a test tube. Then 5 ml of Lowry et al.’s reagent was added and allowed to stand for 15 min at room temperature. Then 0.5 ml of Folin–Ciocalteu reagent was added and the contents were vortexed vigorously and incubated at room temperature for 30 min. This reaction gave purple chromogen in the presence of protein. The protein content was estimated spectrophotometrically (UV-1800 UV-Vis spectrophotometer, SHIMADZU Corporation, Tokyo, Japan) at 750 nm wavelength. The bovine serum albumin (1–10 mg) was used as standard. The results of total protein concentration were expressed as mg/ml of supernatant.
Estimation of TBARS
The tissue TBARS content was estimated as described by the method of Niehaus and Samuelsson. 44 Briefly, 0.5 ml tissue homogenate was mixed with 2.5 ml of (10%, w/v) trichloroacetic acid (TCA) solution and placed in a boiling water bath for 15 min. The test tubes were cooled at room temperature and centrifuged at 1500 rpm for 10 min. Then 2 ml of each supernatant was transferred to a test tube containing 1 ml of TCA (5% w/v) – TBA (0.38% w/v) – HCl (0.25 N) mixture. Test tubes were again placed in a boiling water bath for 15 min and cooled at room temperature. This reaction gave pink chromogen in the presence of malondialdehyde. The absorbance changes in the tissue samples were estimated spectrophotometrically at 535 nm wavelength. The 1,1,3,3-tetramethoxypropane (1–10 nmol) was used as standard. The results of TBARS were expressed as nmol of malondialdehyde/mg of protein.
Estimation of reduced GSH content
The tissue GSH content was estimated using the method described by of Beutler et al. 45 Briefly, 0.5 ml tissue homogenate was mixed with 2 ml of (0.3 M) disodium hydrogen phosphate solution and 0.25 ml of (0.001 M) freshly prepared DTNB solution. This reaction gave yellow chromogen in the presence of GSH. The absorbance changes in the tissue samples were estimated spectrophotometrically at 412 nm wavelength. The GSH (10–100 μmol) was used as standard. The results of GSH were expressed as μg of GSH/mg of protein.
Experimental protocol
Nine groups, each comprising six Swiss mice were employed in the present study.
Group I (normal control)
Mice were not subjected to any surgical procedure and were kept for 14 consecutive days. The nociceptive threshold was assessed at different time intervals: 0, 1, 3, 7 and 14 days. All of the animals were sacrificed on the fourteenth day and biochemical estimations (i.e. total protein, TBARS and reduced GSH level) were performed in the sciatic nerve tissue sample.
Group II (sham control)
Mice were subjected to the surgical procedure to expose the right sciatic nerve without any nerve ligation to differentiate the nerve mediated pain from CCI group. Behavioral and biochemical evaluations were carried out as described in group I.
Group III (CCI)
Mice were subjected to the surgical procedure to expose and ligate the right sciatic nerve. The procedure was described in an earlier section. Behavioral and biochemical evaluations were carried out as described in group I.
Group IV (CCI + vehicle)
After performing the chronic constriction injury of the sciatic nerve, vehicle (25 ml/kg of 0.5% carboxy methyl cellulose) was administered orally for 14 consecutive days from day 0. Behavioral and biochemical evaluations were carried out as described in group I.
Group V and VI (ramipril per se and pregabalin per se)
Mice were subjected to the administration of ramipril (4 mg/kg, p.o.) and pregabalin (10 mg/kg, p.o.) for 14 consecutive days respectively. Behavioral and biochemical evaluations were carried out as described in group I.
Group VII and VIII (ramipril treatment)
Mice were subjected to the administration of ramipril (2 and 4 mg/kg, p.o.) for 14 consecutive days respectively. Behavioral and biochemical evaluations were carried out as described in group I.
Group IX (pregabalin treatment)
Mice were subjected to the administration of pregabalin (10 mg/kg, p.o.) for 14 consecutive days respectively. Behavioral and biochemical evaluations were carried out as described in group 1. This group served as positive control for the study of neuropathic pain.
Statistical analysis
All of the results were expressed as mean ± standard deviation (SD). Data obtained from behavioral tests were statistically analyzed using two-way analysis of variance (ANOVA) followed by Bonferonni’s post hoc analysis, applied by using Graph pad prism Version 5.0 software. The data of tissue biomarkers, i.e. TBARS and GSH levels, were analyzed using one-way ANOVA -followed by Tukey’s multiple range tests, applied for post hoc analysis by using Sigmastat Version 3.5 software. A probability value of P < 0.05 was considered to be statistically significant.
Results
Effect of ramipril on plantar test
CCI of the sciatic nerve resulted in a significant development of thermal hyperalgesia as indicated by a decrease in right hind paw withdrawal threshold when compared to the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced decrease in the thermal nociceptive pain threshold in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced thermal hyperalgesia (Figure 1).

Effect of ramipril on plantar test (paw heat hyperalgesia).
Effect of ramipril on pin prick test
CCI of sciatic nerve resulted in a significant development of mechanical hyperalgesia as indicated by an increase in the percentage withdrawal of right hind paw when compared with the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced increase in the mechanical nociceptive pain threshold in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced mechanical hyperalgesia (Figure 2).
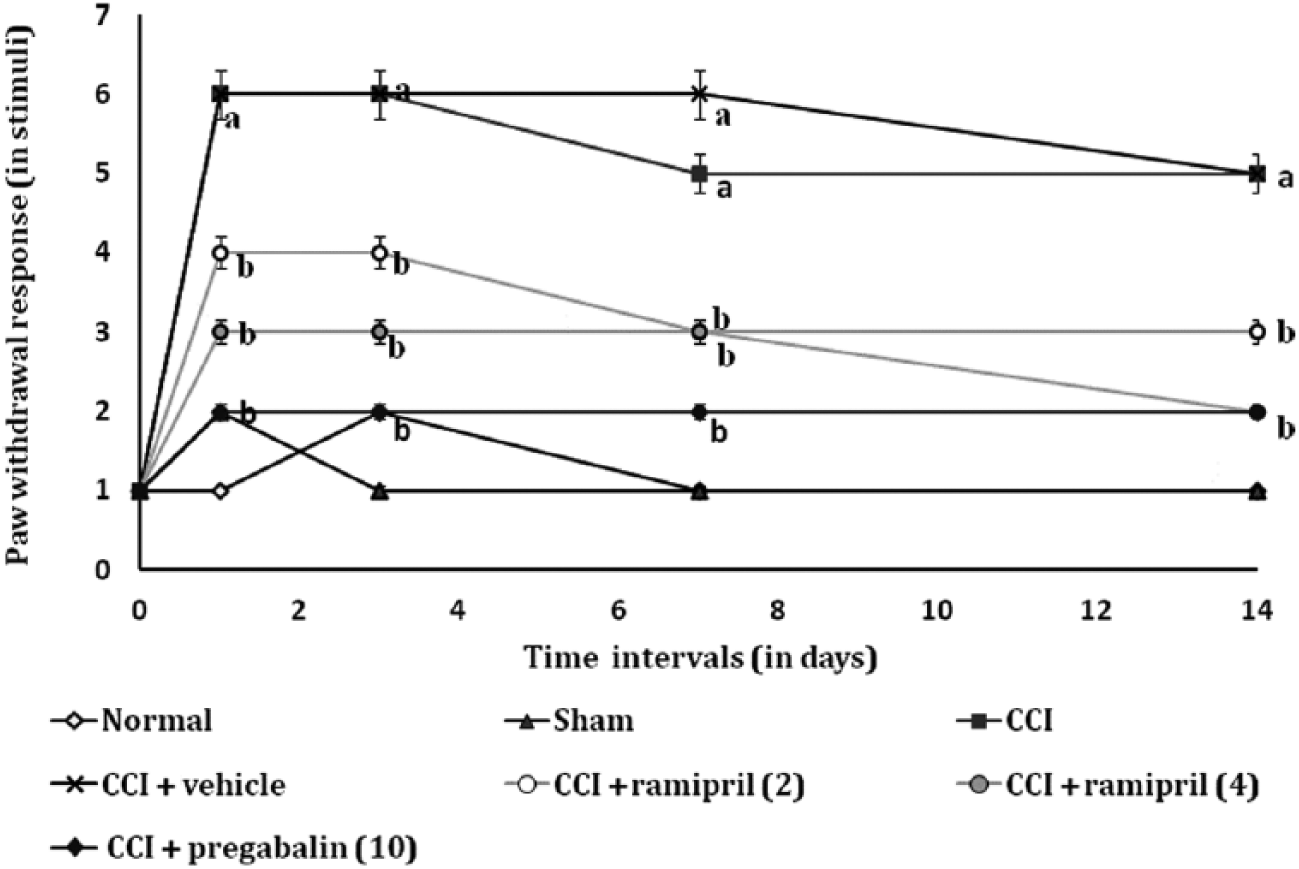
Effect of ramipril on pin prick test (paw mechanical hyperalgesia).
Effect of ramipril on tail flick test
CCI of the sciatic nerve resulted in a significant development of thermal hyperalgesia as indicated by a decrease in tail withdrawal threshold when compared with the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced decrease in the thermal nociceptive pain threshold in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced thermal hyperalgesia (Figure 3).
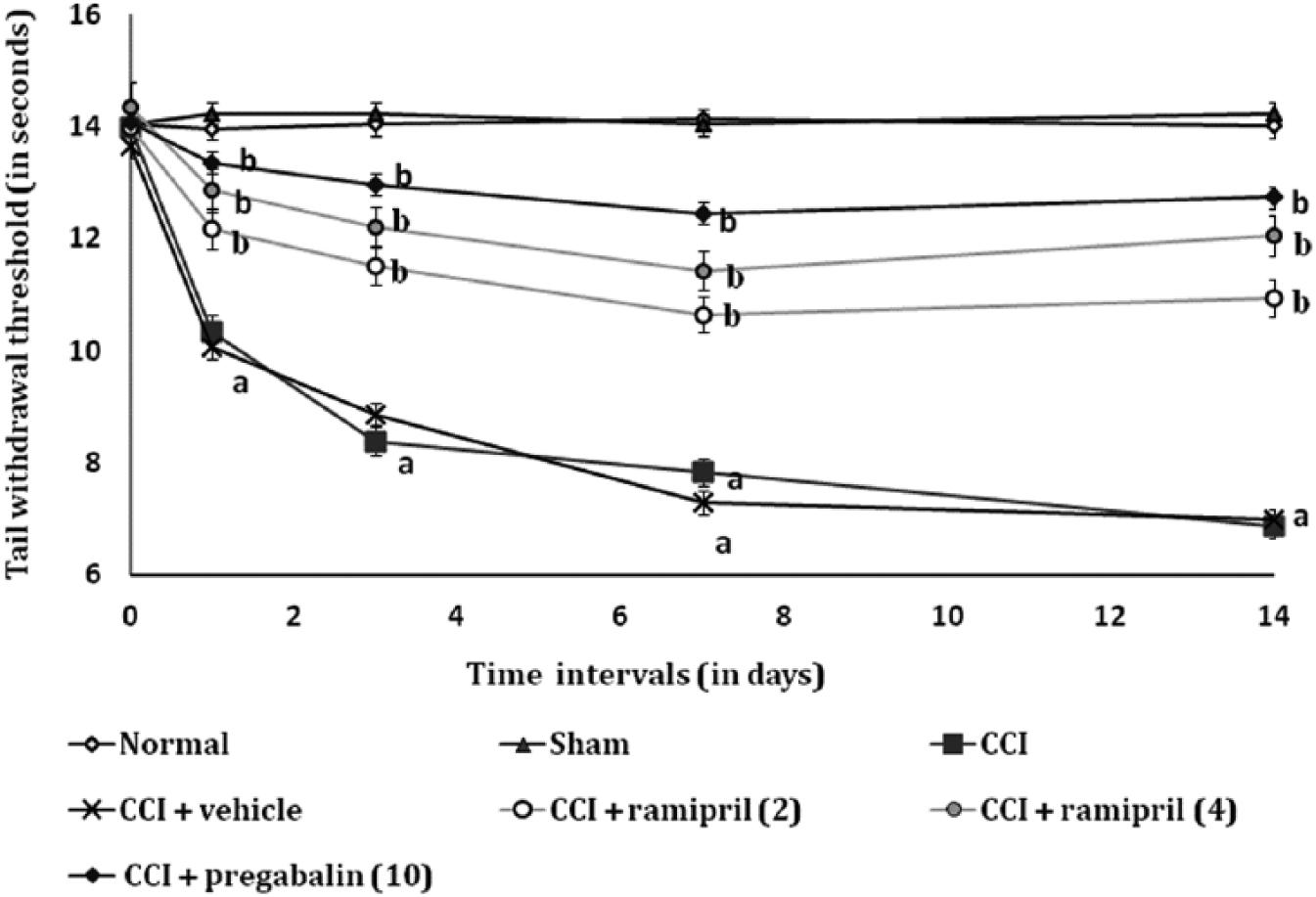
Effect of ramipril on tail flick test (tail heat hyperalgesia)
Effect of ramipril on tail pinch test
CCI of sciatic nerve resulted in a significant development of mechanical hyperalgesia as indicated by an increase in the number of dislodgements of Heffner’s clamp from the tail when compared with the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced increase in the mechanical nociceptive pain threshold in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced mechanical hyperalgesia (Figure 4).

Effect of ramipril on tail pinch test (tail mechanical hyperalgesia).
Effect of ramipril on rota rod test
CCI of the sciatic nerve resulted in a significant development of motor in-coordination as indicated by a decrease in the fall off time from the rotating rod when compared with the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced decrease in the motor impairment in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced motor in-coordination (Figure 5).

Effect of ramipril on rota rod test (motor in-coordination).
Effect of ramipril on tissue biomarker changes
CCI of the sciatic nerve resulted in a significant increase in TBARS and decrease in reduced glutathione content when compared to the sham control group. Administration of ramipril (2 and 4 mg/kg, p.o.) attenuated CCI-induced changes of tissue biomarkers in a dose-dependent manner. Treatment of pregabalin also produced similar effects. However, vehicle and ramipril per se treated group did not show any significant (P < 0.05) changes in CCI-induced tissue biomarker changes (Table 1).
Effect of ramipril on CCI induced oxidative stress marker changes.

CCI: chronic constriction injury; TBARS: thiobarbituric acid reactive substances; GSH: reduced glutathione.
Digits in parentheses indicate dose in mg/kg.
Data were expressed as mean ± SD, n = 6 mice per group.
P < 0.05 vs. sham control group.
P < 0.05 vs. CCI control group.
Discussion
In the present study, CCI of the sciatic nerve in mice has produced a significant increase in thermal and mechanical hyperalgesia and impairment of motor in-coordination along with various biochemical alterations (i.e. rise in TBARS and decrease in reduced glutathione content). Administration of ACE inhibitor, i.e. ramipril (2 and 4 mg/Kg, p.o.) significantly attenuated CCI-induced behavioral and biochemical changes.
The chronic constriction injury model is the most commonly employed neuropathic pain model36,46 which clinically resembles complex regional pain syndrome.42,47 Various pathological mechanisms and targets are explored in the development of CRPS in humans as well as experimental animals. 9 A recent study reported that RAAS plays a critical role in the management of neuropathic pain disorders,17,19,21 as RAAS activation is due to the peripheral nerve injury associated with sympathetic outflow in secondary and tertiary neurons at spinal cord and dorsal root ganglion.11,14 RAAS mediated release of excess angiotensin II is a major culprit in the development of neuropathic pain sensitivity via angiotensin I and angiotensin II receptors.17,20 Recent reports show that angiotensin I receptor blockers, i.e. telmisartan, produce an ameliorative effect against CCI-induced neuropathic pain. 48 The same study revealed that the molecular mechanism of telmisartan includes its direct or secondary effects (partial agonist of peroxisome-proliferator-activated-receptor-gamma) independent of angiotensin I receptor blockade actions. 48 The formation of angiotensin II is due to activity of ACE on angiotensin I. Angiotensin II potentially induces the formation of free radicals associated lipid peroxidation16,49 and neuro-inflammation. 50 ACE inhibitors are well documented in the production of anti-inflammatory action by reduction of angiotensin II formation.51,52 Angiotensin II exerted an anti-nociceptive effect at the variable time points but generally it has high pain sensitivity, because the pain attenuating effect is in a diurnal pattern (24 h light and dark cycles). At phasic pain (dark cycle), it exerted an anti-nociceptive effect, whereas in tonic pain (light cycle), it exerted weak circadian fluctuation of the number of pain responses in mice. 53 Angiotensin II has pain control effects; 54 in contrast, angiotensin II causes the nociceptive pain which is ameliorated by angiotensin 1–7 (AT1-7) peptide via inhibition of Mas receptors associated p 38 MAPK phosphorylation in mice. 55 However, the clinically available medicine for AT2R antagonists is not available yet. AT2R antagonists may be available for chronic pain management in the future. 56 In addition, the rationale of ACE inhibitors increases the pain sensitivity, because it not only produces inhibition of ACE activity, but also inactivates the opioid peptide [Leu5]enkephalin in the rat, 57 whereas the treatment of ramipril reported to have beneficial effects on glucose-fed induced sensory polyneuropathy in rats via down regulation of pro-nociceptive kinin B1 receptors. 58
Furthermore, Ang III as well as Ang IV is biologically active and formed from angiotensin II via ACE activity. But the effect of these have not yet been fully elucidated for the cardiovascular, renal system and neuropathic pain.59,60 The rise in the level of angiotensin II and their receptors, i.e. angiotensin I receptor and angiotensin II receptor, is responsible in the pathogenesis of neuropathic pain in experimental animals and in humans.19,20,61 Angiotensin IV is a metabolite of angiotensin II, which acts on specific angiotensin IV receptors, mainly responsible for improving cognitive dysfunction. 62 The role of angiotensin IV remains to be explored in neuropathic pain disorders. ACE inhibitors are responsible for the reduction of blood pressure and neuroprotective action. 52 The elevation of blood pressure has been reported to produce a reduction in pain sensitivity. The over activation of the RAAS is responsible for the rise in blood pressure, but the role in neuropathic pain has not been explored yet. In the present study, the peripheral nerve injury leads to activate supra-spinal sympathetic tone which in turn causes over activation of the RAAS and releases pro-nociceptive peptides (i.e. angiotensin II). In addition, treatment of ACE inhibitor (Delapril) in spontaneously hypertensive rats (SHR), induces the pain sensitivity and decreases sympathetic tone via two preliminary actions: (a) ACE inhibition; and (b) decrease the endogenous opioid peptide, 63 whereas the treatment of ramipril improves the autonomic control by reduction of sympathetic tone but is not reported to have an effect on endogenous opioid peptide levels. 64 Ramipril is one of the major drugs used as an ACE inhibitor, which also produces neuroprotection by free radical scavenging and anti-inflammatory actions.65,66 The present result also revealed neuroprotective action by the attenuation of CCI-induced rise in TBARS and decrease in reduced glutathione content along with various peripheral and central pain parameters. Ramipril is a prodrug (active form is ramiprilat) for ACE inhibition. It is a lipophilic drug that crosses the blood–brain barrier. However, it does not act on somatic ACE (membrane bound) for degradation of central neuropeptides. 67 Ramiprilat penetrates from the blood into the CSF which inhibits the activity of soluble ACE at the spinal cord region.68,69 Therefore, neuropathic pain parameters have assessed on the peripheral site (paw pain sensitivity response) as well as in the central site (tail pain sensitivity response).
It may be concluded that ramipril has the potential ameliorating role in CCI induced thermal and mechanical hyperalgesia in mice. This action is due to the potential of anti-oxidant and anti-inflammatory actions via reduction of angiotensin II. Therefore, ACE inhibition may be a newer potential target in the management of neuropathic pain disorders.
Footnotes
Acknowledgements
The authors are thankful to the Akal Toxicology Research Centre, a unit of Akal College of Pharmacy & Technical Education, Mastuana Sahib, Sangrur-148001, Punjab (India) for supporting this study and providing technical facilities for this work.
Conflict of interest
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.