
Abstract
Myocardial infarction (MI) induces cardiac remodeling. This may increase the susceptibility of the infarcted heart to subsequent ischemic events. While chronic angiotensin II blockade is cardioprotective post-MI, the acute effects of angiotensin II in ischemia-reperfusion injury (IR) remains unclear. In the present study, we tested whether angiotensin II administration altered recovery of left ventricular (LV) function to IR in hearts from sham and MI rats. Echocardiography, LV pressure-volume relationships, and IR performance were established in subsets of sham (N = 27) and MI hearts (N = 41). IR was conducted in red-cell-perfused Langendorff hearts (60 minutes of low-flow ischemia; 30 minutes of reperfusion) during vehicle or angiotensin II infusions (10−7 M). MI hearts were dilated and had reduced fractional shortening and blunted systolic elastance (p < 0.05). Despite systolic dysfunction in MI, functional recovery to IR was similar to sham. Angiotensin II significantly worsened IR performance in sham (p < 0.05), but not MI. The effect of angiotensin II on in vitro cardiomyocyte survival under various pH conditions was also tested. Acidosis increased cardiomyocyte death and angiotensin II potentiated this effect. We conclude that IR performance is similar between sham and MI hearts and that MI hearts are resistant to angiotensin II-induced cardiac dysfunction in response to IR.
Introduction
Myocardial infarction (MI) increases the predisposition to heart failure and is subsequently a major health concern among industrialized societies. 1 Following MI, the heart undergoes prominent structural remodeling in the form of progressive left ventricular (LV) dilation and regional wall hypertrophy. In parallel with these structural changes, the metabolic phenotype is likewise altered. For example, the normal heart relies to a large extent on fatty acid oxidation to meet its metabolic demands, and this metabolic profile shifts toward an enhanced reliance on glucose oxidation in MI, heart failure, and LV hypertrophy.2–4
Humoral signaling is directly linked to myocardial metabolism. In this context, the renin-angiotensin system (RAS) is a critical consideration in the management of ischemic heart disease and significantly contributes to the pathophysiologic sequelae in MI.1,5–9 Angiotensin II signals through two angiotensin II receptor subtypes, i.e. type 1 (AT1) and type 2 (AT2) receptors. The AT1 receptor is responsible for much of the physiologic actions of angiotensin II in the heart. While the adult cardiovascular system shows low expression of the AT2 receptor, its expression can be altered with increased loading states like MI.7,10 Myocardial AT1 receptor density has been shown to increase after MI and myocardial ischemia.6,9,11 Genetic polymorphisms of angiotensin-converting enzyme (ACE) have also been shown to increase the risk for MI. 5 Chronic treatment with ACE inhibitors or specific angiotensin II receptor antagonists has been shown to attenuate LV dilation, heart failure, and ischemia-reperfusion (IR) injury. 8 Thus while there appears to be little question that chronically blocking the RAS after MI provides significant therapeutic benefit, what is less clear is the potentially important role for the RAS in acute ischemic syndromes, such as post-MI cardiogenic shock.3,9,12
The AT1 receptor is a seven-transmembrane, G protein-coupled receptor that is also capable of signaling through G protein-independent pathways. Ligand binding of angiotensin II to the AT1 receptor activates the second messengers inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). These second messengers subsequently promote inotropy by increasing sarcolemmal ICa2+ and [Ca2+]I, and by alkalinizing cardiomyocytes via protein kinase C (PKC) activation of the Na+/H+ exchanger.7,13–16 The role of AT2 receptors is less clear, but they have been shown to act in vasodilation, in part through nitric oxide-cGMP signaling. 7
We and others have shown that angiotensin II improves inotropy in normal rat hearts during conditions of acidosis. 14 Given that ischemia also acutely blunts myofilament Ca2+sensitivity and contractility, at least in part by inducing intracellular acidosis,14,17 we hypothesized that angiotensin II may alkalinize cardiomyocytes and be functionally protective for the post-MI heart during conditions of IR. We tested this hypothesis in a red cell-perfused, Langendorff isovolumic preparation under low-flow ischemia followed by reperfusion. This model well mimics several aspects of common coronary syndromes in which the myocardium is hypoperfused but still functional. We also tested how acidosis and angiotensin II agonism affects in vitro neonatal rat cardiomyocyte (NRCM) survival under normal pH and during acidotic conditions. Our results suggest that IR performance is similar between sham and MI hearts, with angiotensin II worsening IR performance and cardiomyocyte survival in sham, but not MI hearts.
Methods
MI induction
The infarcted rat model used in this study was modified from that previously described. 18 Briefly, male Wistar rats (Charles River) were anesthetized (50 mg·kg−1 sodium pentobarbital), intubated and ventilated with a rodent ventilator (Harvard apparatus). The chest was opened by an anterolateral thoracotomy between the fifth and sixth ribs. A 5-0 silk suture was passed under the left anterior descending coronary artery (LAD) 2 mm inferior to the left atrium and tied. Occlusion was confirmed by a whitish pallor of the LV. The thorax was sutured shut with a 2-0 silk suture and all animals recovered for four weeks (n = 41). Sham-operated animals underwent the same procedure without ligation of the LAD (n = 27). All animals were treated in accordance with institutional guidelines and those established by the American Physiological Society.
Echocardiography
Echocardiography was used to establish in vivo LV function in a subset of sham and MI animals as previously described. 19 Briefly, animals were lightly sedated with xylazine (10 mg·kg−1) and ketamine (50 mg·kg−1). Images were acquired with the animals in the left lateral decubitus position (HP 5500 Sonos) with a 12-MHz transducer. Measurements of LV end-systolic and end-diastolic internal dimensions (LVEDD) and fractional shortening were acquired from M-mode imaging of the parasternal short-axis view. Values were determined by averaging the measurements of at least three consecutive cardiac cycles.
Langendorff-isolated hearts
Langendorff-isolated hearts were perfused as previously described. 18 Briefly, rats were anesthetized with 50 mg·kg−1 of sodium pentobarbital. Hearts were extracted, weighed and mounted on a perfusion cannula in a Langendorff fashion. The red blood cell perfusate consisted of bovine red blood cells suspended in a Krebs-Henseleit buffer at a final hematocrit of 40%. The Krebs-Henseleit buffer contained in millimoles per liter: NaCl 118, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, NaHCO3 26.6, glucose 5.5, lactate 1, palmitic acid 0.4, heparin 60 mU· ml−1, and 4 g % bovine serum albumin (Sigma Chemical Co., St. Louis, MO). The perfusate free ionized Ca2+ concentration was measured and adjusted to 1 mmol· l−1 Ca2+. Gentamycin (0.2 mg·dl−1 ) was added to the perfusate before it was gassed with 20% O2, 3% CO2, and 77% N2, to achieve a PO2 of 100–140 mm Hg and a pH of 7.35–7.4. A small apical cannula was inserted into the LV to clear Thebesian drainage. A second cannula was inserted into the pulmonary artery for collection of venous effluent. An LV balloon was inserted into the left ventricle through the left atrium to measure LV pressures during pacing at 5 Hz both for pressure volume loops and IR tolerance.
LV pressure-volume relationships:
A subgroup of sham (n = 13) and MI hearts (n = 20) underwent a pressure volume challenge over a physiologic range of LV end-diastolic pressures.18,20 Peak systolic pressure and LV developed pressure were measured throughout the Starling challenge, which consisted of increasing LV balloon volume in 50 µl increments with a Hamilton syringe. Over the linear response range, the delta LV systolic pressure/volume was established for each animal to indicate the systolic elastance as previously described. 20 Group means for absolute systolic elastance were generated and also expressed in relative terms (normalized to noninfarcted, viable myocardium).
IR
A second subgroup of sham (n = 14) and MI hearts (n = 21) underwent IR in the presence and absence of angiotensin II. In these studies hearts were perfused in a Langendorff mode and allowed to equilibrate for 30 minutes at constant coronary flow (2 ml/min). Following equilibration, hearts were then infused with vehicle or a final concentration of angiotensin II (10−7 M) for 10 minutes via a flow pump connected to the perfusion cannula. Low-flow ischemia was then induced for 60 minutes, such that the coronary flow was reduced by 85% of baseline to 0.3 ml/min. Hearts were then reperfused for 30 minutes at the baseline level of coronary flow. LV performance and coronary perfusion pressure (CPP) were measured continuously throughout the protocol. Myocardial oxygen consumption (MVO2: μmol/g wet tissue/min) was calculated from the product of O2 concentration (mM) difference between influent and pulmonary artery effluent and the coronary flow rate (ml/min). 21
Post-mortem assessment
Hearts were arrested with potassium chloride and fixed by retrograde perfusion with 4% paraformaldehyde at a perfusion pressure of 60 cm H2O for 10 minutes with the LV balloon volume set to yield an end-diastolic pressure of 5 mm Hg. Hearts were then suspended in paraformaldehyde for at least 72 hours. After fixation, the left and right ventricles were separated and weighed. The LV length was then established by measuring the longitudinal length from the aortic valve to the inner aspect of the LV apex as previously described. 18 The left ventricle was dissected into four equivocal, transverse sections from apex to base and embedded in paraffin. Sections of 4 µm from each segment were stained with Masson’s trichrome and infarct size was determined as the mean percentage of epicardial and endocardial circumference occupied by scar tissue for the four sections as previously reported. 18
Cell culture
NRCMs from Sprague Dawley rats were isolated from pups via Trypsin digestion. The cells were plated on a 96-well plate at a density of 3 × 104 cells/well and cultured in vitro in a humidified 5% CO2 incubator with Dulbecco’s Modified Eagle Medium (DMEM) medium supplemented with 10% fetal calf serum and the antibiotics penicillin and streptomycin until confluent. At 90% confluence, NRCMs were stimulated with 100 nM angiotensin II (Sigma-Aldrich) or vehicle for two hours at pH = 7.4 or pH = 7.0 (HCl added to DMEM). Viability and cytotoxicity were assessed under normal and low pH conditions using the Multi-Fluor Multiplex Cytotoxicity assay from Promega (Madison, WI). Media glucose and lactate concentrations were assessed with a YSI 2300 Analyzer (Yellow Springs, OH).
Statistics
All data are reported as mean ± SEM. Group differences for physical characteristics and echocardiography between sham and MI hearts were assessed with unpaired T tests. Group differences for IR tolerance were assessed with analysis of variance (ANOVA) with repeated measures followed by T tests when the ANOVA was significant. Group differences for LV pressure volume relationships were also compared with ANOVA followed by unpaired T tests at specific LV volumes. Group differences for cell culture studies were compared using T tests. Data were analyzed with Microsoft Excel and SPPS (Version 20). Statistical significance was set at an alpha level of 0.05 for all analyses.
Results
Physical characteristics
As Table 1 illustrates, body weight was similar in sham and MI animals. Rats with MI had significantly greater heart weights, heart weight/body weights, LV weights, and LV/body weight ratios vs sham (p < 0.05). The average infarct size was 24 ± 2%. Rats with sham operation had an LV length of 1.48 ± 0.04 cm, while rats with MI had an LV length of 1.53 ± 0.02 cm (p = NS). The right ventricular weight was similar between sham (0.26 ± 0.04 g) and MI (0.30 ± 0.02 g; p = NS).
Physical characteristics of rats with sham operation and myocardial infarction.
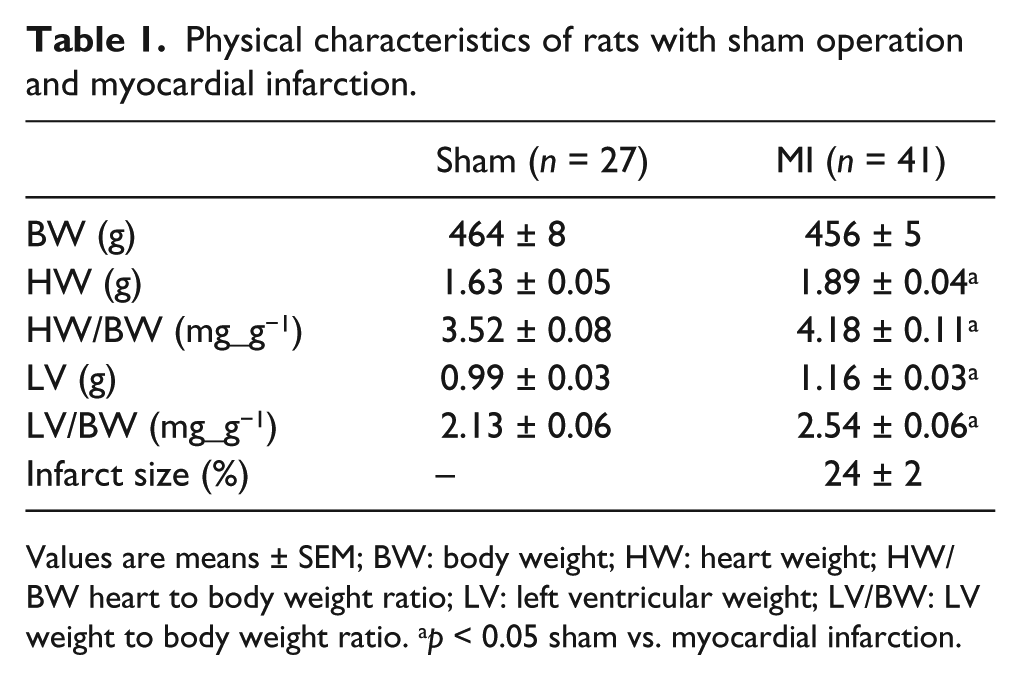
Values are means ± SEM; BW: body weight; HW: heart weight; HW/BW heart to body weight ratio; LV: left ventricular weight; LV/BW: LV weight to body weight ratio. ap < 0.05 sham vs. myocardial infarction.
Echocardiography
As Figure 1 depicts (panels (a) and (b)), the LV internal dimension at end diastole and the LV internal dimension at end systole both increased in MI vs sham hearts (p < 0.05). Fractional shortening was also lower in MI hearts (Panel (c)).
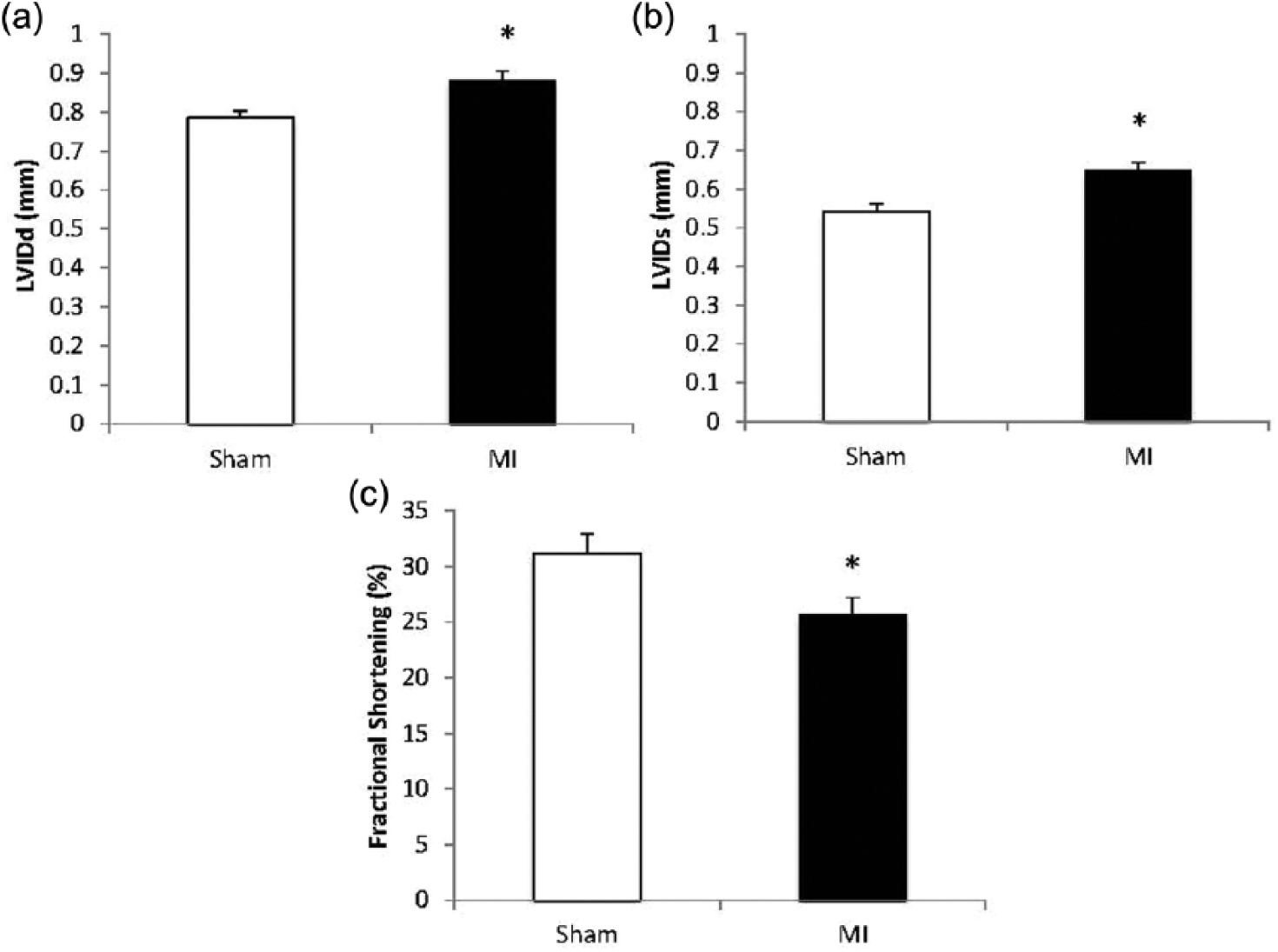
Echocardiography. The figure illustrates that rats with myocardial infarction (MI) showed an increase in left ventricular internal diameter during diastole (LVIDd; panel (a)) and systole (LVIDs; Panel (b)) relative to sham hearts. Fractional shortening was also decreased in MI vs sham. *p < 0.05 between groups.
Starling relationships
As Figure 2 illustrates (panels (a) and (b)), the LV systolic pressure volume relationship was blunted and the LV end-diastolic pressure volume relationship was shifted rightward in MI. The LV-developed pressure was lower in MI vs sham throughout the volume range (Panel (c)). The systolic elastance, i.e. the slope of the end systolic pressure-volume relationship was significantly larger in sham than in MI (p < 0.05) (Panel (d)), and this difference was mitigated when systolic elastance was normalized to viable, noninfarcted myocardium (Panel (e)).
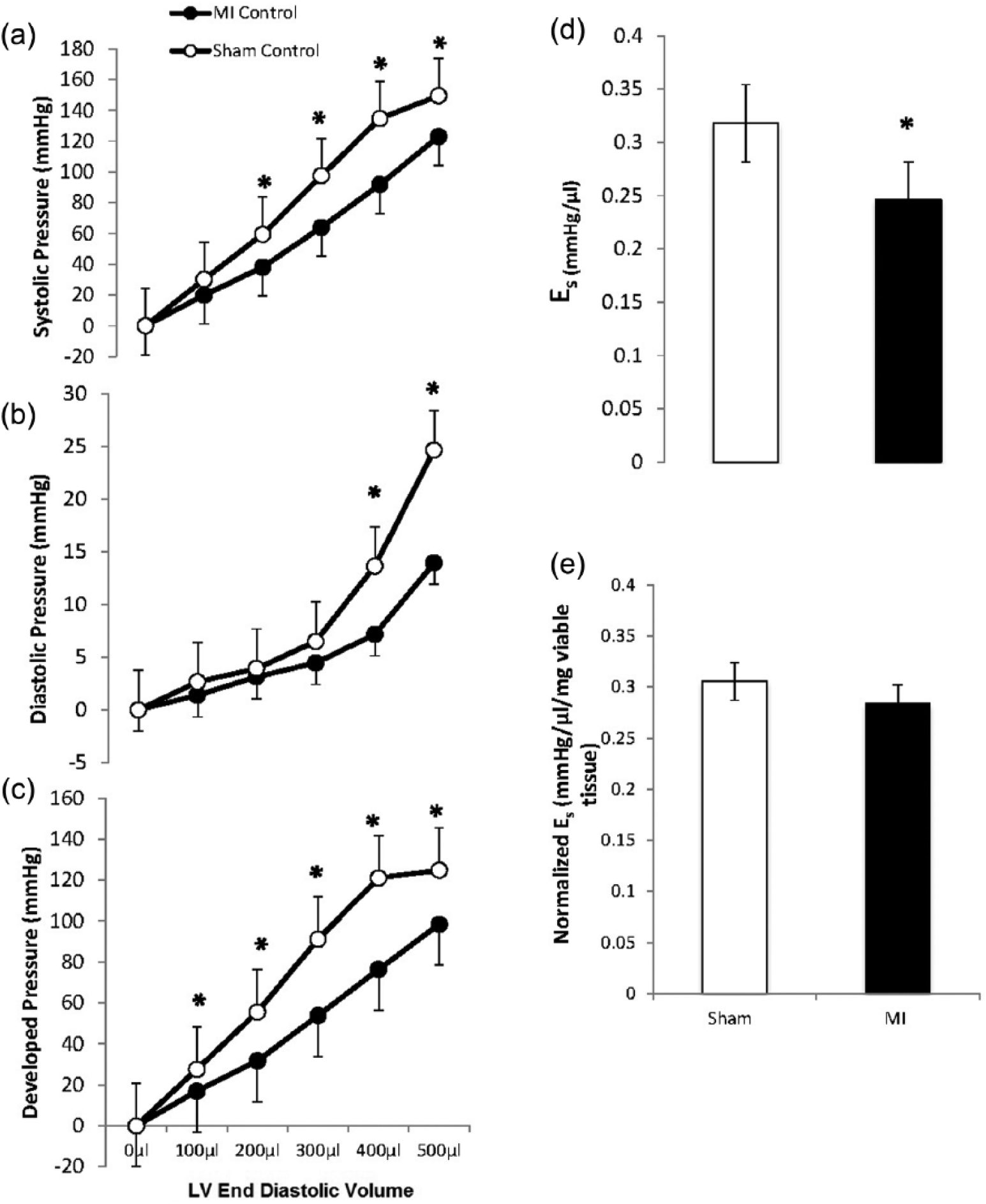
Left ventricular pressure volume relationships. The left ventricular balloon volume was incrementally increased over a range of 500 µl in Langendorff-perfused sham and myocardial infarction (MI) hearts. Left ventricular (LV) systolic pressures (panel (a)) and LV-developed pressures (Panel (c)) were lower in MI vs sham hearts. At higher LV balloon volumes, LV diastolic pressure was lower in MI hearts (Panel (b)). The systolic elastance was reduced in MI hearts (Panel (d)) but this difference was negated when systolic elastance was normalized to noninfarcted viable tissue mass (Panel (e)). *p < 0.05 between groups.
IR
LV performance at baseline was similar between sham and MI Langendorff hearts (Table 2). As Figure 3 illustrates (panels (e) and (j)), angiotensin II infusion increased CPP during normoxic, baseline perfusion relative to vehicle (sham baseline: 62 ± 4 mmHg vs sham Ang II: 70 ± 4 mmHg, p < 0.05; MI baseline: 71 ± 4 mmHg vs MI Ang II 80 ± 4 mmHg, p < 0.05) without affecting LV performance in either sham or MI. Also as Figure 3 shows (panels (a)–(n)), LV performance during IR (as indexed by LV-developed pressure, LVEDP, dP/dt max, dP/dt min, CPP, MVO2 and pH) was similar between sham and MI hearts. Angiotensin II infusion did not affect LV functional performance during ischemia in either sham or MI, but it increased CPP relative to baseline and attenuated recovery (% of LV developed pressure) during reperfusion in sham hearts. During early reperfusion, angiotensin II did not alter MVO2 or coronary venous effluent pH of sham or MI hearts. The functional recovery to IR was not different between angiotensin II and vehicle conditions in MI.
Baseline Langendorff performance.
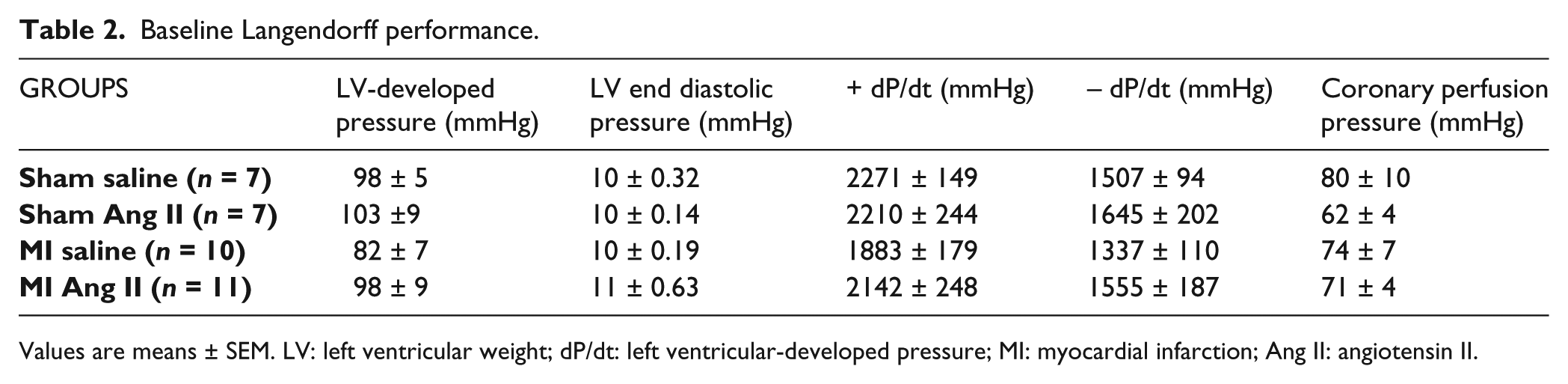
Values are means ± SEM. LV: left ventricular weight; dP/dt: left ventricular-developed pressure; MI: myocardial infarction; Ang II: angiotensin II.
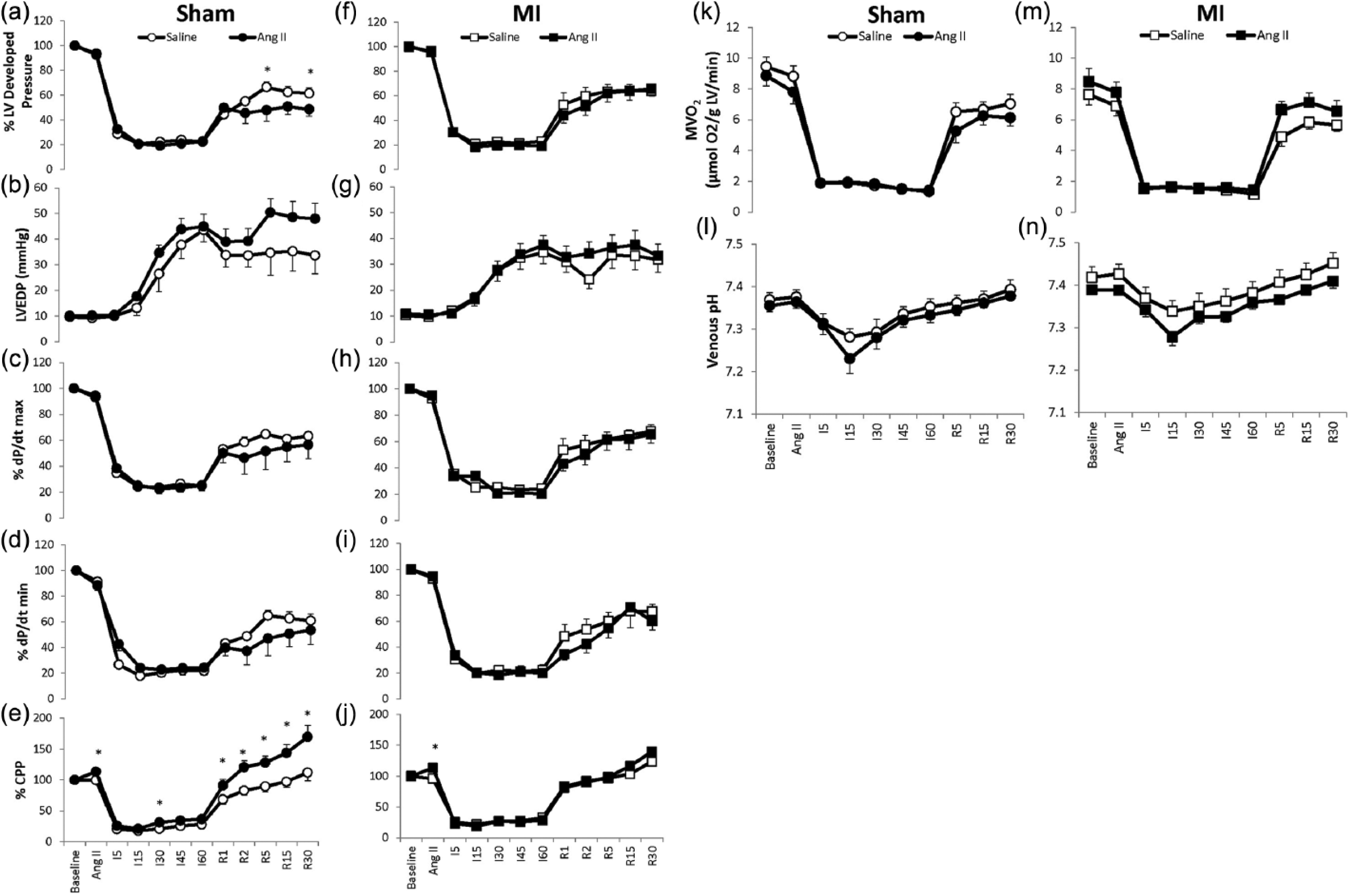
Ischemia-reperfusion tolerance. Hearts were perfused in a Langendorff mode and allowed to equilibrate for 30 minutes at constant coronary flow (2 ml/min). Following equilibration, hearts were then infused with vehicle or a final concentration of angiotensin II (10−7 M) for 10 minutes via a flow pump connected to the perfusion cannula. Low-flow ischemia (I) was then induced for 60 minutes, such that the coronary flow was reduced by 85% of baseline to 0.3 ml/min. Hearts were then reperfused (R) for 30 minutes at the baseline level of coronary flow. As the figure illustrates, myocardial infarction (MI) hearts showed a similar functional recovery to ischemia reperfusion performance as sham. Angiotensin II infusion further worsened percentage recovery of LV-developed pressure (Panel (a)) and increased coronary perfusion pressure (CPP) (Panel (e)) in sham but not MI in the later stages of reperfusion. *p < 0.05 vs respective saline vehicle infusion.
Cell culture
We also tested how angiotensin II affected NRCMs during normal pH and under acidotic conditions. As Figure 4(a) illustrates, survival of NRCMs was reduced under low pH conditions compared to normal pH. NRCMs cultured with angiotensin II did not provide protection from cell death, and instead potentiated NRCM death both under normal pH (p = 0.09) and acidotic conditions (p < 0.05). Acidosis also attenuated glucose consumption and blunted lactate appearance without an observed angiotensin II effect. These data suggest that alterations in glucose consumption are not the reason by which angiotensin II exacerbated cell death.
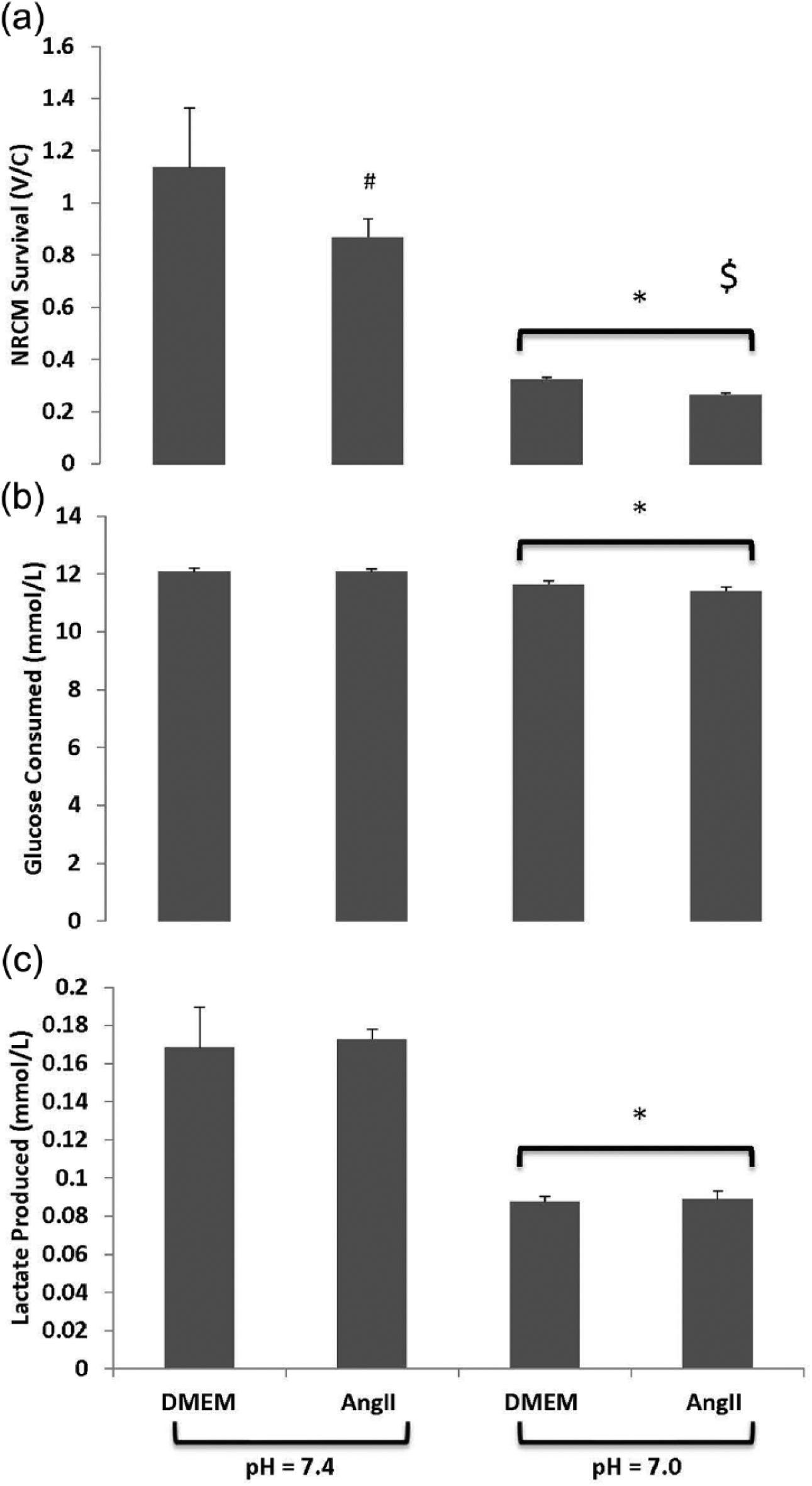
Cell culture. Neonatal rat cardiomyocytes (NRCMs) were isolated and plated on a 96-well plate at a density of 3 × 104 cells/well. NRCMs were stimulated with 100 nM angiotensin II or vehicle for two hours at pH = 7.4 or pH = 7.0. Cell survivability (Panel (a)) shows that survival of NRCMs was reduced under low pH conditions vs normal pH. NRCMs cultured with angiotensin II did not provide protection from cell death, and instead potentiated NRCM death both under normal pH (#p = 0.09) and acidotic conditions ($ indicates p < 0.05). Acidosis also attenuated glucose consumption (Panel (b)) and blunted lactate production (Panel (c)) without an observed angiotensin II effect. *p < 0.05 normal pH vs acidosis.
Discussion
In this study we tested the hypothesis that the alkalizing actions of angiotensin II may be functionally protective for the post-MI heart during acute conditions of IR. IR tolerance was similar between MI and sham hearts despite the phenotypical changes observed in MI hearts, i.e. LV dilation and systolic dysfunction. Interestingly, exogenous angiotensin II infusion impaired cardiac performance during IR in sham but not MI hearts. Using cultured NRCMs, we found that angiotensin II potentiated the effect of acidosis to increase cardiomyocyte cell death. We conclude that angiotensin II does not provide functional protection to the infarcted heart when diffuse myocardial ischemia is present. Angiotensin II instead hastened IR LV dysfunction and enhanced cell death in noninfarcted hearts and cultured cardiomyocytes, respectively.
The model of coronary ligation used in the present study produced typical and expected post-MI remodeling of the heart. 18 MI hearts were dilated and hypertrophied and showed a reduction in systolic performance. LV internal diameter on echocardiography was increased with MI, and there was a rightward shift in the LV end diastolic pressure volume relationship. Systolic function was also compromised in MI hearts, as evidenced by a reduction in fractional shortening and a lower systolic elastance. It is interesting to note that when systolic elastance was normalized to the mass of viable LV tissue, the differences between sham and MI became insignificant. These data suggest that force per viable muscle mass was not compromised at this stage of the post-MI remodeling process. It is also important to note that Langendorff-derived systolic pressure was nearly equivocal between sham and MI hearts when compared at similar LV end diastolic pressures.
The interrelationship between myocardial metabolism and the development of post-MI heart failure is not fully established. While a decrease in overall myocardial energy production has been shown to occur in severe LV dysfunction, this finding is less consistent in nonfailing, infarcted hearts 2 . Substrate shifts with MI, such as an increased reliance on glucose utilization and a reduction in fatty acid oxidation, have often been reported 2 . Moreover, declining creatine phosphate/adenosine triphosphate (PCr/ATP) ratios are correlated with poor LV function.2,21–24 Thus on the basis of shifts in substrate utilization and PCr metabolism, one might hypothesize that IR performance would be impaired in MI hearts relative to noninfarcted hearts. Results from the present study do not support this hypothesis. Instead, our data show that LV functional performance during IR was nearly identical between MI and sham hearts. Beyond the similar functional IR phenotype observed in sham and MI, oxidative metabolism as indexed by MVO2 was also similar between MI and sham. These findings are consistent with a recent report suggesting that infarcted, nonfailing hearts do not necessarily show oxidative metabolic defects. 2
The metabolic milieu may be critical in determining how angiotensin II affects cardiac function. This is well illustrated in a paper by Horn et al, who showed that chronic quinapril therapy preserved both LV function and high-energy phosphate metabolism in post-MI hearts during normoxic conditions. 3 However, quinapril therapy elicited lower ATP concentrations and worsened LV performance during reoxygenation following hypoxia. These data suggest that the RAS may be compensatory in oxygen deprived conditions like ischemia. It is well known that shifts in pHi can affect myocardial inotropy, with alkalosis increasing inotropy via augmented troponin C– Ca2+ sensitivity. Since pHi is known to be reduced during ischemia, and previous work has shown that angiotensin II can induce intracellular alkalinization, we hypothesized that angiotensin II may preserve cardiac function during IR by causing a relative alkalosis.13–16, 25
In the present study, angiotensin II infusion increased coronary resistance both in sham and MI hearts during baseline normoxic perfusion conditions. Concomitant with the increase in coronary perfusion pressure with angiotensin II infusion at baseline, LV performance fell relative to vehicle conditions in both groups. During low-flow ischemia, angiotensin II did not alter cardiac performance, MVO2, or pH in either sham or MI relative to vehicle conditions. Yet during reperfusion, coronary resistance was significantly increased with angiotensin II infusion in sham hearts; and percentage recovery for LV-developed pressure in sham was likewise impaired relative to vehicle conditions. Based on coronary effluent pH measures during IR, we did not detect significant angiotensin II pH effects in the present study. Moreover, coronary venous effluent pH was similar between MI and sham. The finding that angiotensin II exacerbated IR dysfunction in sham is consistent with the potential pro-oxidative stress of angiotensin II. Angiotensin II has been shown to stimulate H2O2 and O2− formation via activation of NAD(P)H oxidases.25–28 However, our finding of exacerbated IR dysfunction in sham hearts is contrary to a prior study by Ford et al., who showed that angiotensin II reduced infarct size but elicited no adverse effects on post-ischemic contractile performance in a buffer-perfused, global no-flow ischemia, working heart model. 29 Differences in experimental paradigms may explain these variable results.
Conversely, angiotensin II did not elicit a detrimental effect in MI hearts during IR. This is surprising given that myocardial angiotensin II receptor density has been shown to increase after MI. 7 Our results instead suggest blunted intracellular angiotensin II signaling in infarcted hearts. Differential agonist or autocrine signaling through AT1 and AT2 in MI may also explain the blunted angiotensin II effect in MI. 30 Yang et al. reported an increase in AT1 receptor expression immediately after IR in isolated rat hearts. 9 AT2 expression has been shown to be increased in cardiac hypertrophy and failing hearts and overexpression of the AT2 receptor has been shown to offer cardioprotection after MI. 12 Thus signaling through an upregulated AT2 with MI may have counteracted the potentially deleterious effects of angiotensin II through AT1, as was observed in sham. Further studies are needed to elucidate the differential mechanisms for angiotensin II signaling between MI and sham hearts during IR.
In isolated NRCMs, acidosis increased cell death and blunted glucose consumption and lactate production. The reduction in glucose consumption during acidosis may have been related to the inhibition of glycolytic enzyme activity known to occur with acidosis, or it may be reflective of fewer living NRCMs. The addition of angiotensin II further increased NRCM cell death during normal pH and acidosis. However, angiotensin II did not alter glucose uptake by NRCMs, suggesting that changes in glucose consumption are not the reason why angiotensin II exacerbated cell death. Interpreted together with the Langendorff functional heart data, our results in NRCMs support the idea that short-term angiotensin II is not beneficial to the heart during metabolic challenges like IR or acidosis.
In summary, we found that IR performance is not altered in MI hearts despite cardiac remodeling and impaired systolic function. Our findings also indicate that angiotensin II infusion during ischemia and reperfusion is functionally deleterious only in noninfarcted hearts, suggesting post-MI compensatory responses blunt the deleterious side effects of angiotensin II. We conclude that angiotensin II is not beneficial when diffuse myocardial ischemia is present, i.e. cardiogenic shock.
Footnotes
Acknowledgements
The authors would like to thank Soeun Ngoy for his technical contributions toward the MI model and the Margulies laboratory for the provision of neonatal cardiomyocytes.
Conflict of interest
None declared.
Funding
This study was supported by the Biobehavioral Research Center, University of Pennsylvania School of Nursing.