
Abstract
Liver cancer kills approximately 800 thousand people annually worldwide, and its most common subtype is hepatocellular carcinoma (HCC), which usually affects people with cirrhosis. Predicting survival of patients with HCC remains an important challenge, especially because technologies needed for this scope are not available in all hospitals. In this context, machine learning applied to medical records can be a fast, low-cost tool to predict survival and detect the most predictive features from health records. In this study, we analyzed medical data of 165 patients with HCC: we employed computational intelligence to predict their survival, and to detect the most relevant clinical factors able to discriminate survived from deceased cases. Afterwards, we compared our data mining results with those obtained through statistical tests and scientific literature findings. Our analysis revealed that blood levels of alkaline-phosphatase (ALP), alpha-fetoprotein (AFP), and hemoglobin are the most effective prognostic factors in this dataset. We found literature supporting association of these three factors with hepatoma, even though only AFP has been used in a prognostic index. Our results suggest that ALP and hemoglobin can be candidates for future HCC prognostic indexes, and that physicians could focus on ALP, AFP, and hemoglobin when studying HCC records.
Keywords
Introduction
Hepatocellular carcinoma (HCC), or hepatoma, is the most common liver cancer and affects millions of people worldwide (14 million just in 2012 1 ). Liver cancer kills approximately 800 thousand individuals worldwide annually.2,3 HCC especially inflicts those with liver cirrhosis, which commonly can be caused by excessive alcohol consumption or viral hepatitis. This kind of cancer is more common among men, between 30 and 50 years of age, and is more frequent in mainland China, Mongolia, South-East Asia, and Sub-Saharan Western and Eastern Africa. 4
Similar to the other types of cancer, hepatocellular carcinoma makes cells reproduce at a higher rate and can make the cells avoid apoptosis.5,6
HCC is usually diagnosed with computed tomography (CT) scan and magnetic resonance imaging (MRI), and its treatment includes invasive procedures, such as surgical resection, liver transplantation, radiofrequency ablation (RFA), arterial catheter-based treatment, systemic therapy, or radioembolization. 7
Analysis of electronic health records of patients diagnosed with hepatoma has become an effective method to forecast prognosis and survival likelihood. Detecting which patients with hepatoma have a high risk of death can be extremely useful to arrange the proper therapy or treatment, and therefore to make more precise efforts to save their lives. However, it is also important to identify patients that have more chances to survive, to avoid them to have invasive treatments such as liver transplantation, surgical resection, or chemotheraphy.
From the 1980s, the scientific community started selecting some factors from these clinical records to generate liver cancer prognostic indexes, in order to classify the patients with HCC and to try to predict their prognosis: the Okuda system, 8 the Cancer of Liver Italian Program (CLIP), 9 the Barcelona Clinic Liver Cancer (BCLC), 10 the GRoupe d’Etude et de Traitement du Carcinoma Hépatocellulaire (GRETCH), 11 tumour-node-metastasis classification scheme, 12 the Chinese University Prognostic Index (CUPI), 13 the Japanese Integrated System (JIS), 14 the estrogen receptor (ER) molecular staging system, 15 and the TNM Classification of Malignant Tumors.16,17
Even if all useful, no consensus on a common, standard, and unified prognostic index has been reached among the medical community, 18 leaving room for the design of alternative indices involving other risk factors and health record features, especially through computerized systems.
In this context, computational methods applied to clinical records of patients diagnosed with hepatocellular carcinoma can be useful in predicting the likelihood of patient survival and in detecting the most relevant survival-related features. Machine learning, especially, is capable of identifying hidden patterns in data, and can provide rankings of risk factors computed automatically.
For these reasons, researchers took advantage of data mining techniques applied to health records several times in the past, especially on data of patients with cancer.19–22
Regarding hepatocellular carcinoma, Tannus et al. 23 employed several traditional statistical methods to analyze data of 247 patients with HCC from Brazil, and compared several hepatocellular carcinoma prognostic systems. Gui et al. 24 applied data mining techniques to a gene expression of 95 samples to detect the genes most related to hepatocellular carcinoma. Also Ye et al. 25 used a similar approach on a gene expression of 67 samples, to predict hepatitis B virus–positive metastatic hepatocellular carcinomas. The study of Yim et al. 26 shows an application of supervised machine learning techniques to radiology data of patients with HCC.
In the present article, we analyze a dataset of 165 patients having HCC (Dataset). After data pre-processing and imputation, we apply several supervised machine learning methods to computationally predict their survival, as a classic binary classification task. Afterwards, we take advantage of the top performing method (Random Forests) to rank the clinical features of the dataset on their predictive power. Finally, we use traditional univariate statistical methods to evaluate the association between each feature and survival, without employing machine learning.
Our survival prediction methods outperform the results obtained by the original dataset authors, 27 and our feature ranking indicates alkaline phosphatase (ALP), α-fetoprotein (AFP), and hemoglobin levels as the most predictive survival factors.
We organized the rest of the article as follows. After this Introduction, we describe the dataset we analyzed (section “Dataset”) and the methods we used (section “Methods”); we then describe the survival prediction results and the medical feature rankings (section “Results”) and discuss their meanings and relevance (section “Discussion”). Finally, we draw some conclusions, describe the limitations and the future developments of the present study (section “Conclusion”).
Dataset
The analyzed dataset contains clinical records of 165 patients diagnosed with hepatocellular carcinoma (HCC), collected at the Centro Hospitalar e Universitàrio de Coimbra in Portugal from 1st January 2008 to 31st December 2013.27,28 Each patient profile contains 50 features, including the 1-year survival (class 0: deceased; class 1: survived) that we use as target in this study.
As mentioned by the original dataset curators, 27 the variables of this dataset have been selected according to the guidelines of European Association for the Study of the Liver—European Organization for Research and Treatment of Cancer (EASL-EORTC). 29
The dataset contains features related to blood tests (AFP, AHT, ALP, ALT, AST, Creatinine, Direct Bilirubin, Ferritin, GGT, Hemoglobin, Iron, Leucocytes, MCV, platelet count, Total Bilirubin, and Total Protein levels), presence of other diseases (Cirrhosis, Diabetes, and Obesity), and personal features, such as Age and Sex (Table 1). The patients include 32 women and 133 men. We report the complete meaning of the dataset features in Table 1. For clarification purposes, we slightly changed some of feature names (Supplemental Material Information).
Meanings and measurement units of each feature of the dataset.
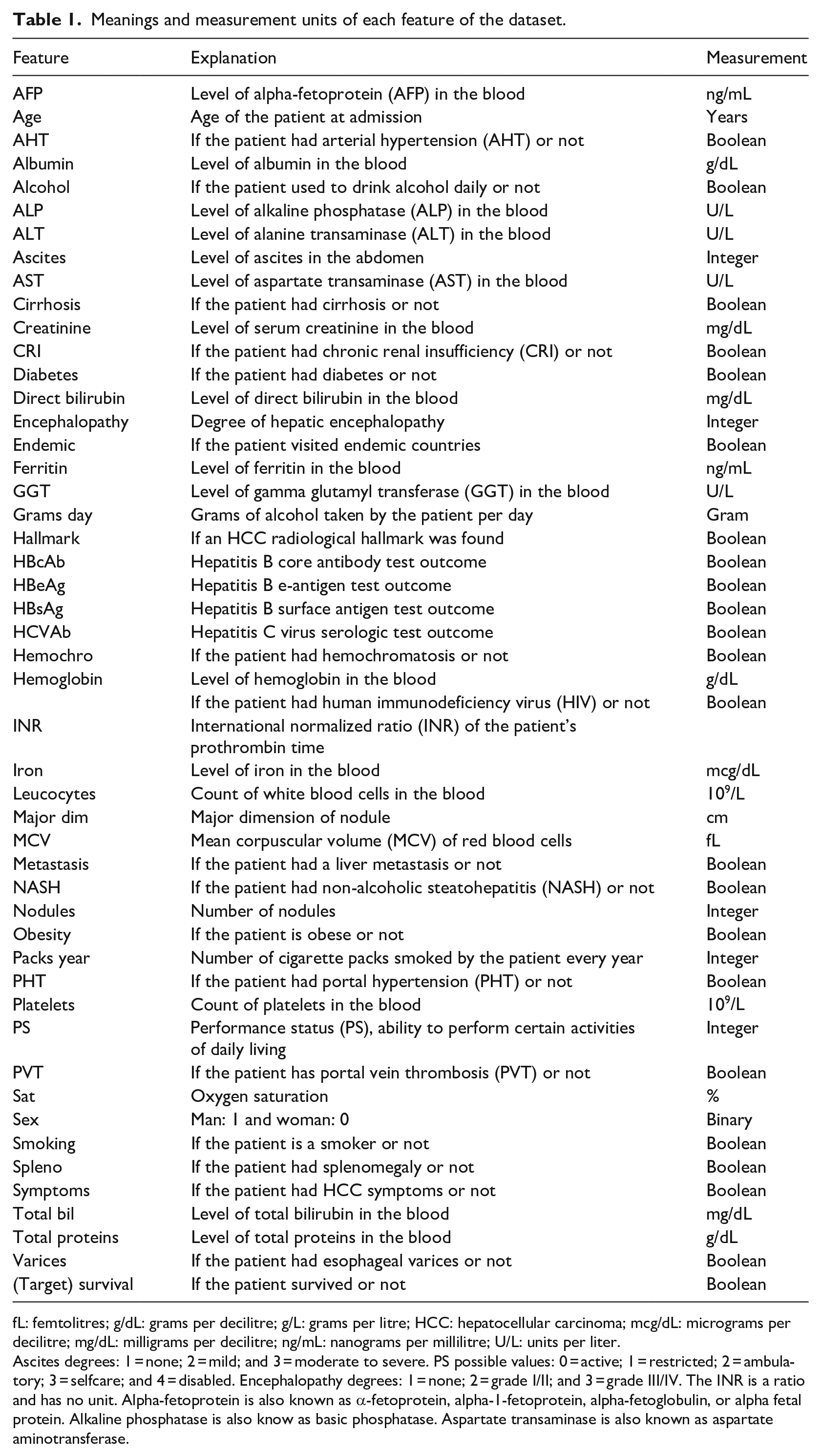
fL: femtolitres; g/dL: grams per decilitre; g/L: grams per litre; HCC: hepatocellular carcinoma; mcg/dL: micrograms per decilitre; mg/dL: milligrams per decilitre; ng/mL: nanograms per millilitre; U/L: units per liter.
Ascites degrees: 1 = none; 2 = mild; and 3 = moderate to severe. PS possible values: 0 = active; 1 = restricted; 2 = ambulatory; 3 = selfcare; and 4 = disabled. Encephalopathy degrees: 1 = none; 2 = grade I/II; and 3 = grade III/IV. The INR is a ratio and has no unit. Alpha-fetoprotein is also known as α-fetoprotein, alpha-1-fetoprotein, alpha-fetoglobulin, or alpha fetal protein. Alkaline phosphatase is also know as basic phosphatase. Aspartate transaminase is also known as aspartate aminotransferase.
A total of 24 clinical factors have binary values (Table 2), while 23 have real values or ordinal category values (Table 3). Most of the clinical features have missing values, with oxygen saturation (Sat) and Ferritin having the maximum percentage of 48.485% missingness (Table 3).
Statistical quantitative description of the binary features.
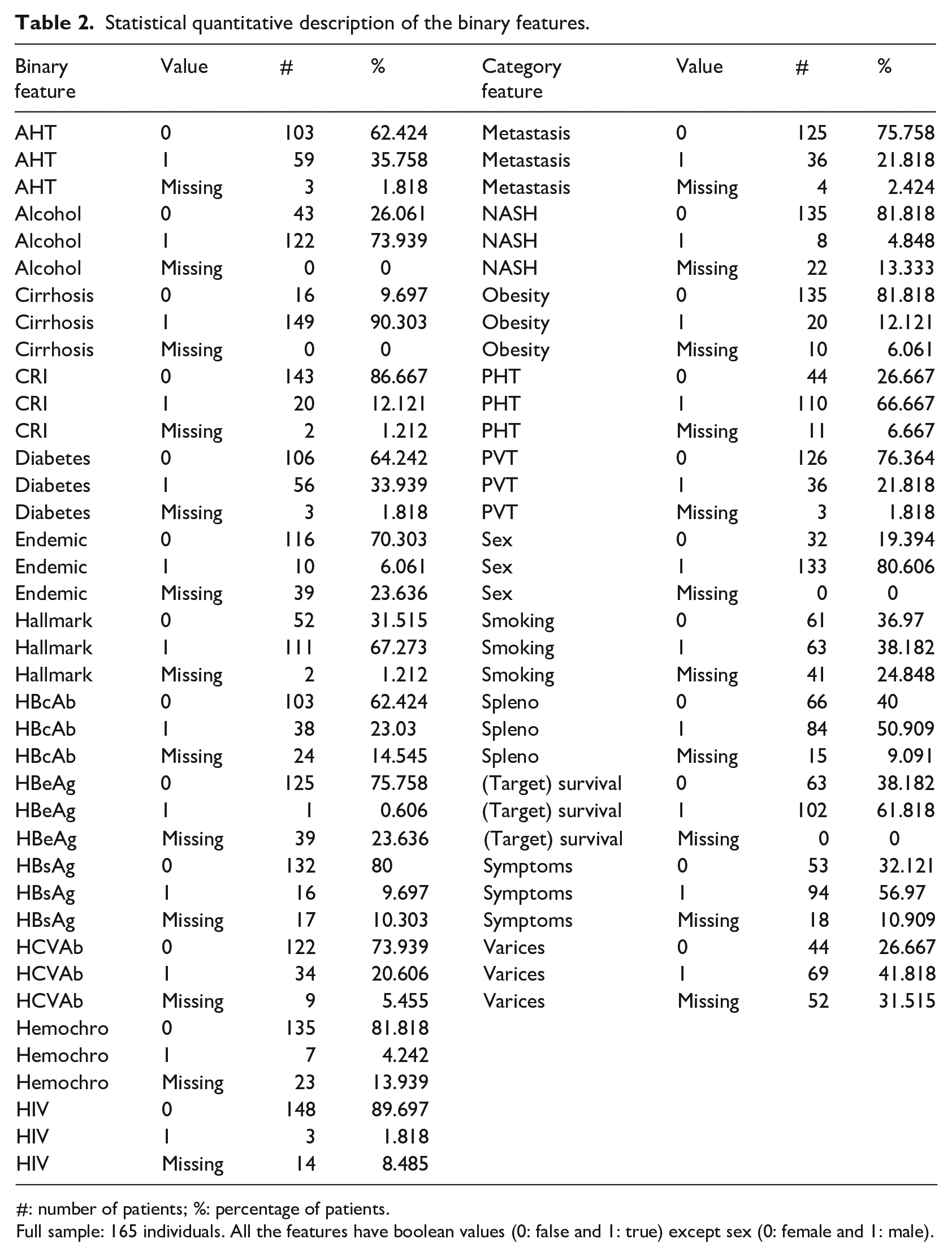
#: number of patients; %: percentage of patients.
Full sample: 165 individuals. All the features have boolean values (0: false and 1: true) except sex (0: female and 1: male).
Statistical quantitative description of the numeric features.
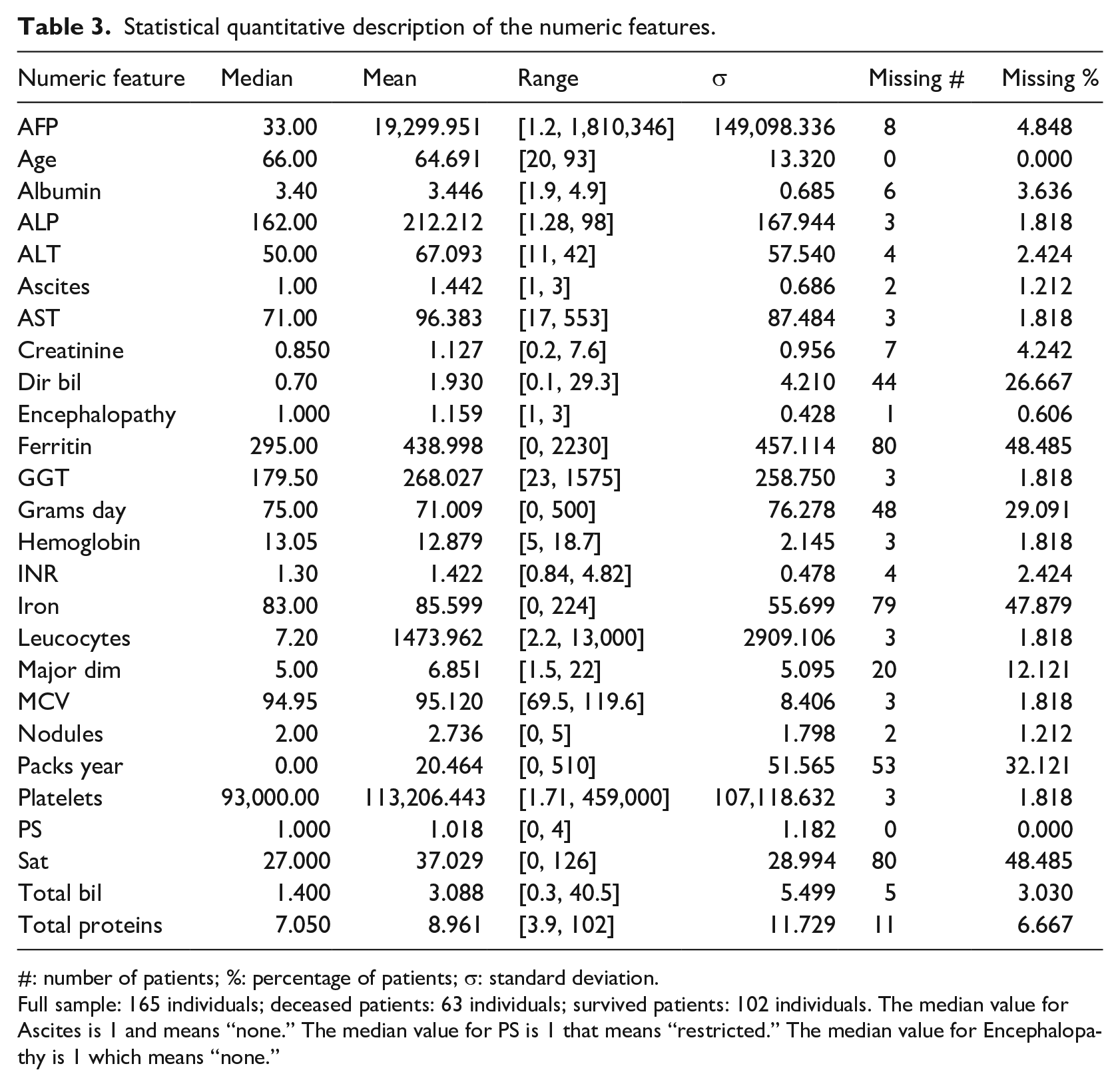
#: number of patients; %: percentage of patients; σ: standard deviation.
Full sample: 165 individuals; deceased patients: 63 individuals; survived patients: 102 individuals. The median value for Ascites is 1 and means “none.” The median value for PS is 1 that means “restricted.” The median value for Encephalopathy is 1 which means “none.”
The dataset is slightly imbalanced toward the positive class: there are 102 survived patients and 63 deceased patients, meaning 61.82% positive data instances and 38.18% negative data instances. The survival feature refers to 1 year after the hospital visit when the clinical data were recorded: if the patient profile has the positive survival label, it means she/he survived after 1 year; if the patient profile has a negative survival label, it means that she/he deceased within 1 year.
More information about this dataset can be found in the original publication by Santos et al. 27 and in the University of California Irvine Machine Learning Repository. 28
Methods
In this section we describe both the methods we used for binary classification and for feature ranking.
Let us consider the now-classical binary classification framework.30,31 Let
Before applying the classification and feature ranking algorithms, data must be pre-processed to be able to handle it and extract useful and actionable information (Data pre-processing and imputation). Let us consider a model (function)
Note also that
In fact, once the model is built based on the different learning algorithms and has been confirmed to be a sufficiently accurate representation of the
Data pre-processing and imputation
Before employing a machine learning algorithm, data needs to be pre-processed.
36
In particular,
In the case of categorical feature space with more than two categories, if the algorithm is not able to handle multi-categorical features (for example, Support Vector Machines and Neural Networks), we will opt for the one hot encoding and we map it in a numerical feature space.
32
Note also that some values of
In this case, if the missing value is in a categorical feature, we introduce an additional category for missing values for that feature. If, instead, the missing value is associated with a numerical feature, we replace the missing value with the mean value of that feature and we introduce an additional logical feature to indicate whether the value of that feature is missing or not for a particular sample.
Algorithms
In this section we briefly recall the four algorithms that we have exploited in this study by pointing out the idea behind them, how to use them, and their hyper-parameters. The selected algorithms represent the most effective algorithms in four families of methods 31 : rule based methods, ensemble methods, kernel methods, and neural networks.
Decision Tree
A binary Decision Tree (DT) 38 belongs to the family of the rule based methods. The DT is a flowchart-like structure in which each internal node represents a test of a feature, each branch represents the outcome of the test, and each leaf node represents an output of the tree. A path from the root to a leaf represents a model rule.
A DT is built with a recursive schema until it reaches its desired depth d, which is the DT hyper-parameter that needs to be tuned during the MS phase. Each node of the DT, starting from the root node, is built by choosing the attribute and the cut that most effectively split the set of samples into two subsets based on the information gain.
The decision trees can handle categorical features, numerical features, and missing values well, and they do not suffer from numerical issues (no normalization of the data is needed).
Random Forests
The Random Forests (RF) 39 belong to the family of the ensemble methods. RF combine bagging to random subset feature selection. In bagging, each tree is independently constructed using a bootstrap sample of the dataset. RF add an additional layer of randomness to bagging. In addition to constructing each tree using a different bootstrap sample of the data, RF change how the classification trees are constructed.
In standard trees, each node is split using the best division among all variables. In a RF, each node is split using the best among a subset of predictors randomly chosen at that node. Eventually, a simple majority vote is taken for prediction. The accuracy of the final model depends mainly on three different factors: how many trees compose the forest, the accuracy of each tree and the correlation between them. The accuracy for RF converges to a limit as the number of trees
There are several hyper-parameters which characterize the performance of the final model: the number of trees, the number of samples to extract during the bootstrap procedure, the depth of each tree, the number of predictors exploited in each subset during the growth of each tree, and finally the weights assigned to each tree. Nevertheless, in common applications, the RF stability to these factors is quite low. 39
Since RF is basically a combination of many DTs, RF can handle categorical features, numerical features, and missing values well, and they do not suffer from numerical issues (no normalization of the data is needed).
Support Vector Machines
The Support Vector Machines (SVM) 40 belong to the family of the kernel methods.
Kernel methods are a family of techniques which exploits the “kernel trick” for distances to extend linear techniques to the solution of non-linear problems. 41 Kernel methods select the model which minimizes the trade-off between the performance, measured with a defined metric (Supplemental Material Information), over the data and the complexity of the solution, measured with different measures of complexities.31,40 Support Vector Machines (SVM), linear SVM (linear) and non-linear SVM (kernel), represent the most known and effective Kernel methods techniques.
The hyper-parameters of the SVM include the kernel, which is usually fixed and is the linear one for SVM (linear) and the Gaussian one for SVM (kernel),
42
the kernel hyper-parameter
SVM cannot handle categorical features directly (therefore, the one hot encoding 32 is needed) and they do suffer from numerical issues and consequently data must be re-scaled (in our case all the numerical features and targets have been scaled to have zero mean and variance equal to one).
Multi-Layer Perceptron Neural Network with Dropout
The Multilayer Perceptron Network with Dropout (MLP)43,44 belongs to the family of the neural networks.
Neural networks are techniques which combine together many simple models of a human brain neuron, called perceptrons, 45 to build a complex network. The neurons are organized into stacked layers, connected together by weights that are learned based on the available data via back-propagation. 46
If the architecture of the neural network consists of only one hidden layer, it is called shallow, while, if multiple layers are staked together, the architecture is defined as deep. From a functional point of view both architectures have the same representation power 47 but in practice, for some applications like natural language processing and image analysis, deep networks outperform the shallow ones.44,48
In our context, where the number of samples and features is limited, it is more reasonable to use a shallow network.43,44 In particular, in this study, we exploited a well known and effective architecture, the MLP, where a single hidden layer is present, we train it with adaptive subgradient methods, and we tuned the following hyper-parameters during the MS phase
44
: the number of neurons in the hidden layer
The MLP, like SVM, fails to handle categorical features directly (consequently, the one hot encoding 32 is needed) and they do suffer from numerical issues and consequently the data must be re-scaled (in our case we exploited the same re-scaling method exploited for SVM).
Handling unbalanced classes
Data available in bioinformatics for binary classification are often strongly unbalanced.49–51 However, most learning algorithms work badly with imbalanced datasets and tend to perform poorly on the minority class and for these reasons several techniques have been developed to address this issue. 33
The first step toward the solution of this problem is to avoid applying the inappropriate evaluation metrics for model generated using imbalanced data.
52
For example, overall accuracy is a very dangerous metric in this context since the more unbalanced is the dataset the more this metric tends to promote models which poorly perform on the minority class. For this reason, in this study we also included other metrics like Matthews correlation coefficient (MCC),
53
The second step toward the mitigation of the effects of having an unbalanced dataset is to modify the algorithm or the data, but currently the most practical and effective method involves the re-sampling of the data to synthesize a balanced dataset. 33 For this purpose we can under- or over-sample the dataset. Under-sampling balances the dataset by reducing the size of the abundant class. By keeping all samples in the rare class and randomly selecting an equal number of samples in the abundant class, a new balanced dataset can be retrieved for further modeling. Note that this method wastes a great deal of information (many samples may not be used). For this reason the oversampling strategy is more often exploited. It tries to balance the dataset by increasing the size of rare samples. Rather than removing abundant samples, artificial synthetized samples are generated (for example by repetition, by bootstrapping, or by synthetic minority). The latter method is the one that we exploited in this paper.
Model selection and error estimation
MS and EE deal with the problem of tuning and assessing the performance of a learning algorithm. 34 Resampling techniques like k-fold cross validation and non-parametric bootstrap are often used by practitioners because they work well in many situations. 57 Other alternatives exist, which represent foundations in the Statistical Learning Theory and give more insight into the learning process. Examples of methods in this last category include the seminal work of the Vapnik-Chervonenkis Dimension, its improvement with the Rademacher Complexity, the theory of compression, the Algorithmic Stability breakthrough, the PAC-Bayes theory, and more recently the Differential Privacy theory. 34
In this work we will exploit the resampling techniques which rely on a simple idea: the original dataset
Then, to select the best combination of the hyper-parameters

where
Then, to evaluate the performance of the optimal model which is

Since the data in
If
Feature ranking
Once we built the models and they showed their effectiveness in predicting the desired quantities, we decided to investigate how these models are affected by the different features used in the model identification phase. We performed this operation to understand if the models also have a foundation which relies on the underlying phenomena or if the model just captures spurious correlations. 35
This procedure is called feature ranking (FR) and allows one to detect if the learned models appropriately take into account the relevant features. We consider relevant features the ones that are known to be important based on the literature or on the knowledge of the experts on the scientific problem.
The failure of the computational model to properly account for the relevant features might indicate poor quality in the measurements or spurious correlations. FR therefore represents an important step of model verification, since it should generate consistent results with the available knowledge of the phenomena under examination.
Since Random Forests was the method which obtained the best results in binary classification (Table 4), we took advantage of this ensemble learning technique to perform the feature ranking.
Results of the survival prediction made with machine learning classifiers.
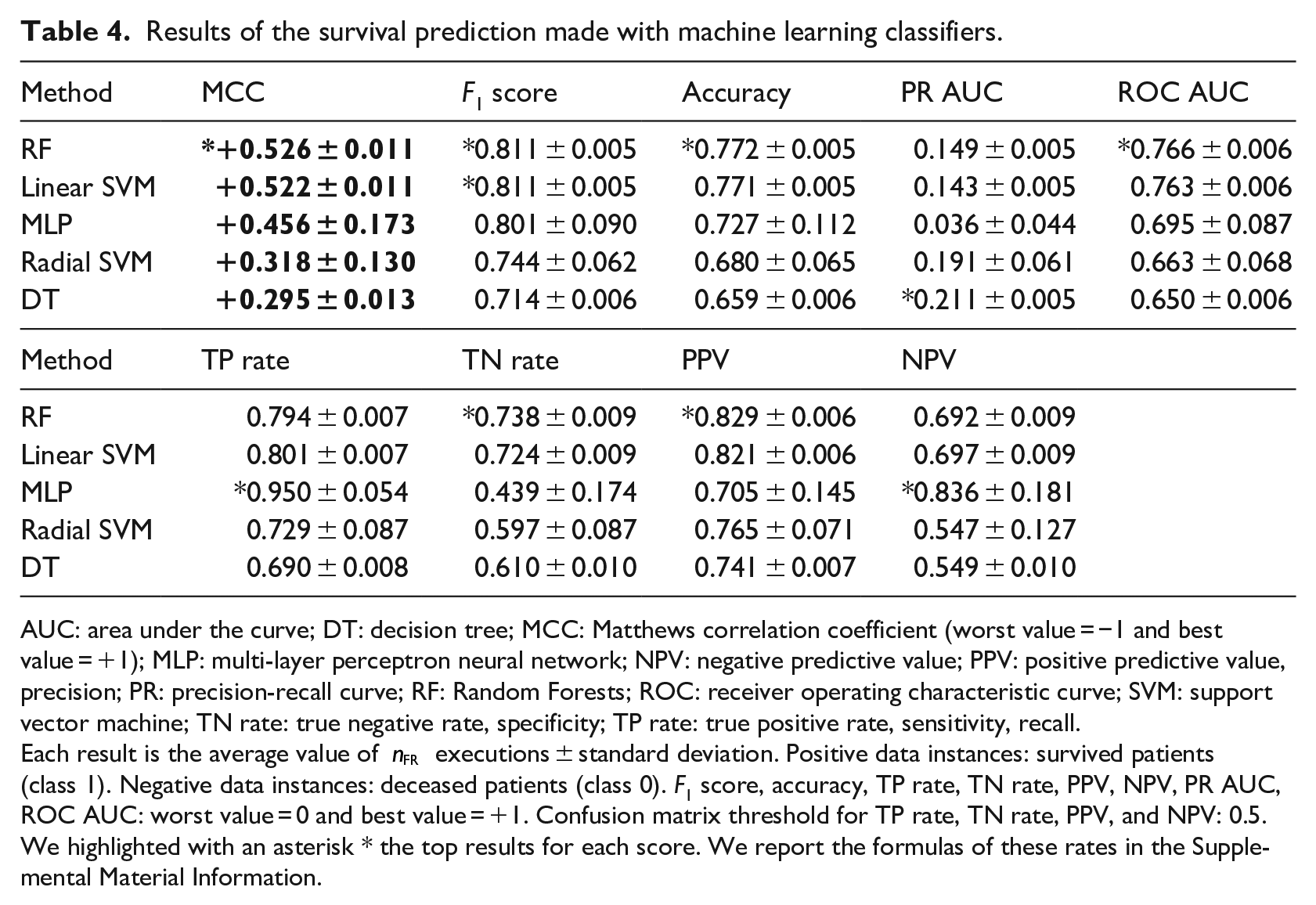
AUC: area under the curve; DT: decision tree; MCC: Matthews correlation coefficient (worst value = −1 and best value = +1); MLP: multi-layer perceptron neural network; NPV: negative predictive value; PPV: positive predictive value, precision; PR: precision-recall curve; RF: Random Forests; ROC: receiver operating characteristic curve; SVM: support vector machine; TN rate: true negative rate, specificity; TP rate: true positive rate, sensitivity, recall.
Each result is the average value of
Random Forests feature ranking
In this context, feature rankings methods based on Random Forests are among the most effective machine learning techniques,58,59 particularly in the context of bioinformatics60,61 and health informatics. 19 Several measures are available for feature importance in Random Forests.
One approach is based on the Gini Importance or Mean Decrease in Impurity (MDI) which calculates each feature importance as the sum over the number of splits (across all trees) that include the feature, proportionally to the number of samples it splits. 62 Another powerful approach is the one based on the Permutation Importance or Mean Decrease in Accuracy (MDA), where the algorithm assesses the importance for each feature by removing the association between that feature and the target. 62 This goal can be achieved by randomly permuting 63 the values of the feature and measuring the resulting increase in error. The method also removes the influence of the correlated features.
In details, for every tree, two quantities are computed: the first one is the error on the out-of-bag samples as they are used during prediction, while the second one is the error on the out-of-bag samples after a random permutation of the values of a variable. These two values are then subtracted and the average of the result over all the trees in the ensemble is the raw importance score for the variable under examination. Both MDI and MDA can be adopted since they can be easily carried out during the main prediction process inexpensively.
Despite the effectiveness of MDI and MDA, when the number of samples is small, these methods might be unstable.64–66 For this reason, in this study, instead of running the Feature Ranking (FR) procedure just once, analogously to what we have done for MS and EE, we sub-sample
Computational pipeline for binary classification and feature ranking
We can recap here the computation pipeline of the analysis with the following steps:
Construction of the dataset described earlier (Data pre-processing and imputation) and pre-process it;
We built a model with each of the algorithms described in Algorithms (DT, RF, SVM (linear), SVM (kernel), and NN). We will handle the unbalanced classes as described in Handling unbalanced classes. We will use the MS strategy described in Model selection and error estimation where we set the number of fold (a) DT: (b) RF: we set (c) SVM (linear): (d) SVM (kernel): (e) NN:
where
For each of the constructed models we reported the results using the EE strategy described in Model selection and error estimation and the confusion matrix metrics together with the standard deviation where we set
We reported the ranking of the features selected by the two feature ranking procedures described in Feature ranking with
Biostatistics univariate tests
After using machine learning for feature ranking, we decided to rank the features by using the results of univariate traditional statistical tests which statistically express the relationship between each clinical factor and survival.
As done by Patrício et al.
20
when analyzing health records of patients with breast cancer, we first applied the Shapiro–Wilk test
68
to each feature to check their distribution. Since the normality assumptions were unmet, we then applied the Mann–Whitney
The Mann–Whitney
The chi-squared test
71
between two variables checks how likely an observed distribution is due to chance. The Mann–Whitney
A low
Results
In this section, we first report and describe the binary classification results we obtained for the survival prediction (section “Survival prediction”), the feature ranking results we obtained through machine learning (section “Machine learning clinical feature ranking”), and the feature ranking results we obtained through traditional statistics tests (section “Biostatistics feature ranking”).
Survival prediction
We listed the survival prediction results in Table 4, by ranking them on the Matthews correlation coefficients. 53 We chose the MCC because it is the only confusion matrix score that generates a high score only if the classifier obtained a high score on the sensitivity, specificity, precision, and negative predictive value.36,56
Our results show that all the methods were able to correctly predict most of the survived patients (positive data instances) and all the methods except MLP were capable of correctly precicting the majority of deceased patients (negative data instances), by obtaining MCC from +0.295 to +0.526 on average (Table 4). Random Forests outperformed all the other methods, by achieving the top MCC,
All methods performed better on recall than on specificity, by obtaining an almost perfect true positive rate of 0.950 (MLP) as top recall and by attaining a top 0.738 true negative rate (RF). We believe this difference is caused by the dataset imbalance, since there are 38.18% negative data instances (deceased patients) and 61.82 positive data instances (survived patients). The high scores of precision and negative predictive value confirm the confidence of our survival prediction. 72
Our results show that the classifiers perform more efficiently when used to predict patients with high chance to decease (TP rate) than patients with high chance to survive (TN rate). This condition results being more advisable to us because, as we mentioned earlier, predicting patients more at risk of death is more urgent.
Machine learning clinical feature ranking
Since Random Forests obtained the best results classifying the survival target (Survival prediction), we decided to employ this method to detect the most predictive features, able to discriminate survived patients from deceased patients. We applied an ensemble learning feature ranking
The results showed ALP, AFP, Hemoglobin, Albumin, and Ferritin as top five clinical factors to distinguish survived patients from deceased patients in both the final Gini ranking and the final accuracy ranking. Regarding the features ranked in the last positions, instead, the two final rankings were discordant. Alcohol, Cirrhosis, Sex, Hallmark, and Obesity resulted being the less relevant factors in the merged ranking (Table 5).
Clinical feature ranking results.
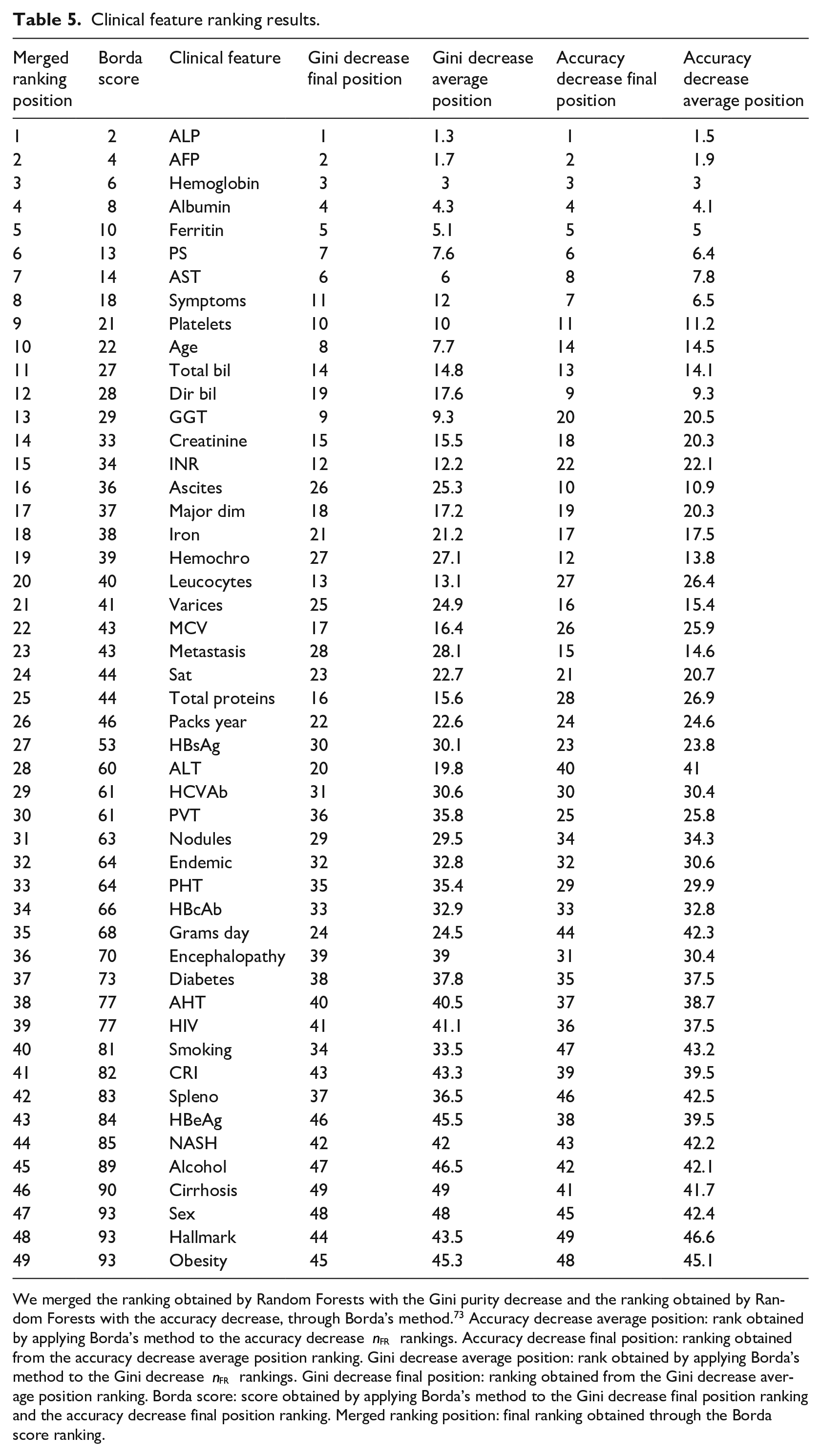
We merged the ranking obtained by Random Forests with the Gini purity decrease and the ranking obtained by Random Forests with the accuracy decrease, through Borda’s method.
73
Accuracy decrease average position: rank obtained by applying Borda’s method to the accuracy decrease
As an example, we report a Gini ranking and an accuracy ranking generated by one of the applications of Random Forests out of
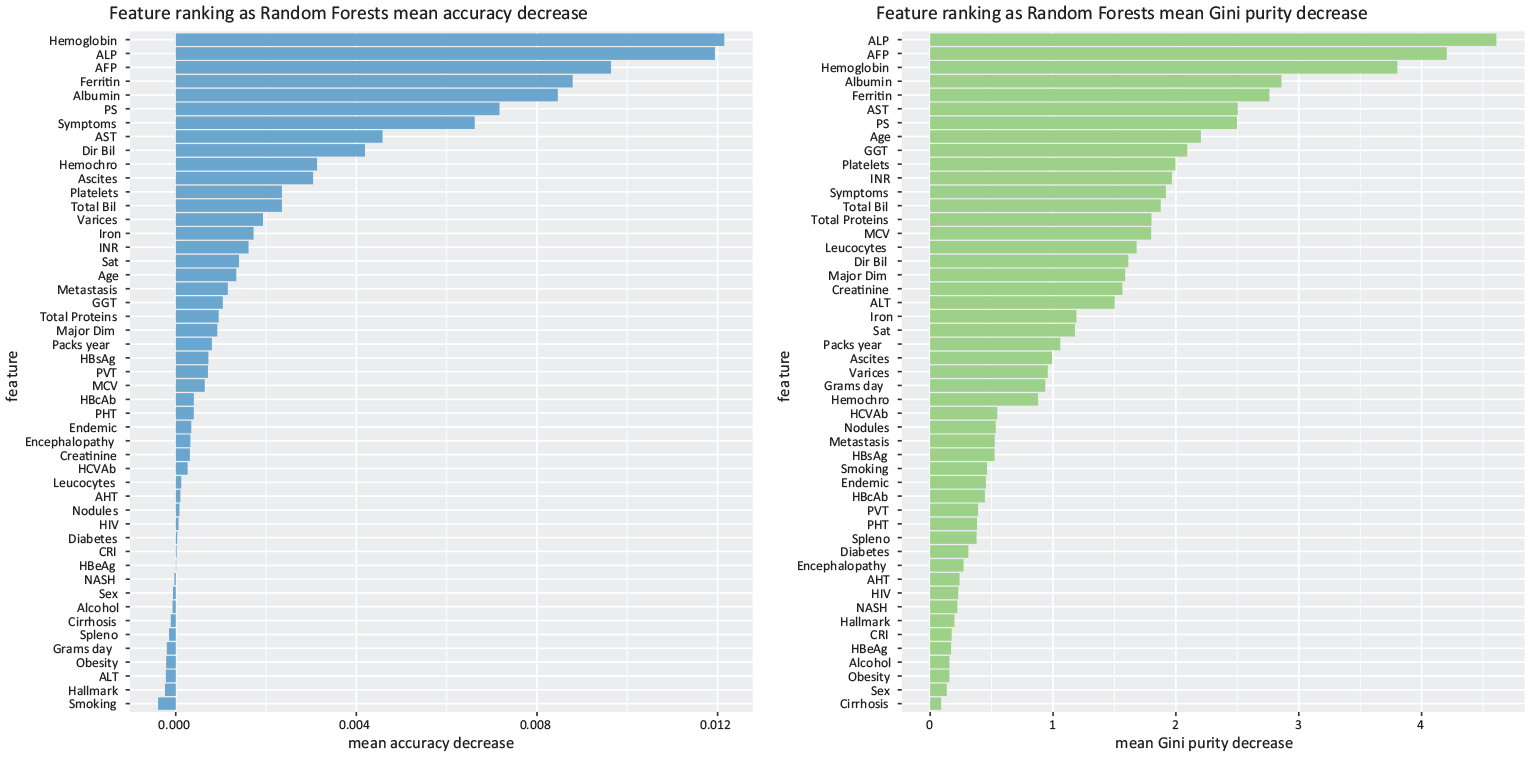
Random Forests feature selection. Indicative example of Random Forests feature selection through accuracy reduction (left) and Random Forests feature selection through Gini impurity (right), obtained in one of the
Biostatistics feature ranking
The Shapiro–Wilk test (Supplemental Table S1) produced a
We applied the Mann–Whitney
Results of the Mann–Whitney
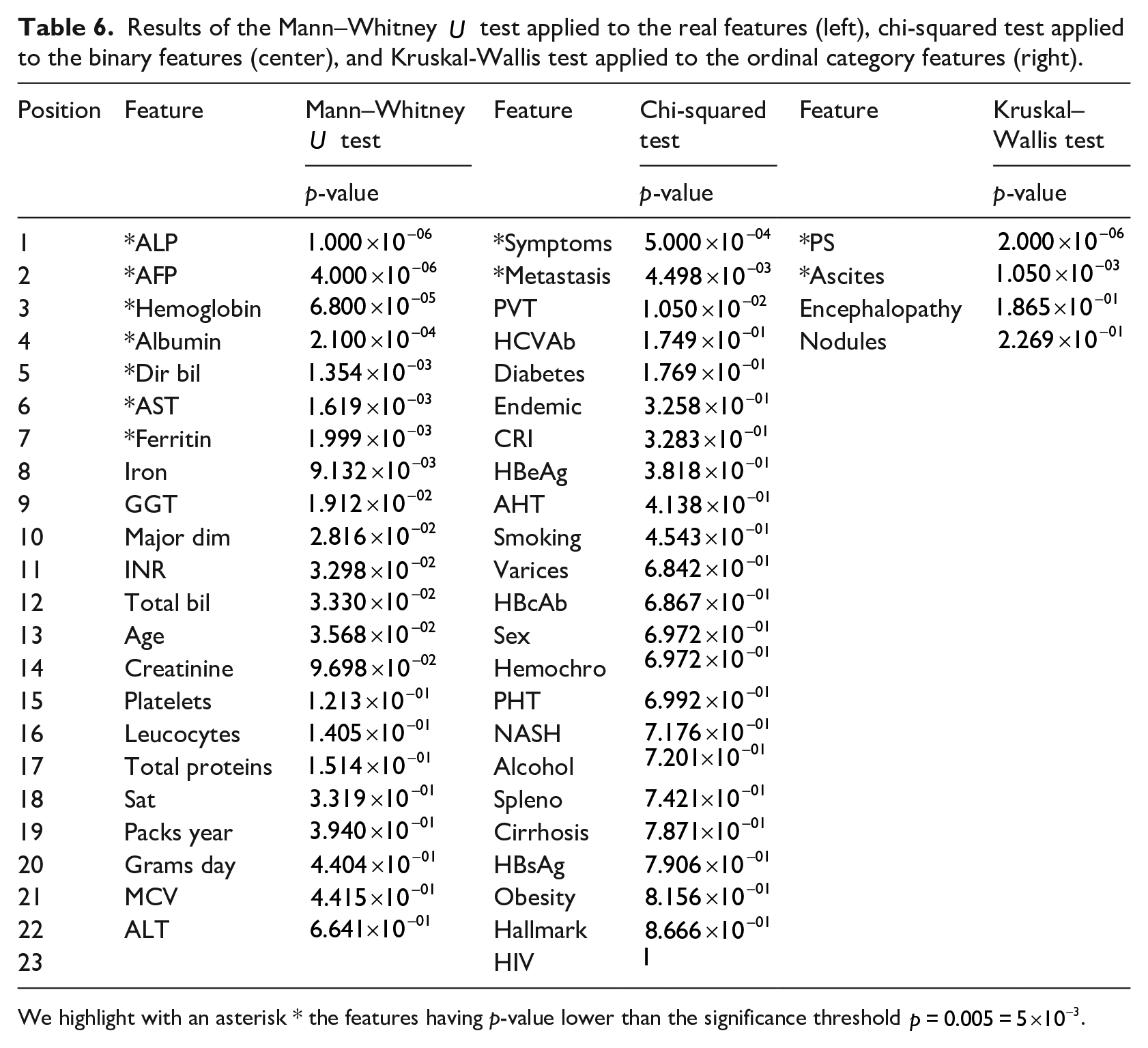
We highlight with an asterisk * the features having p-value lower than the significance threshold
Exection times
We executed our scripts on a Dell Latitude 3540 personal computer running a Linux CentOS 7.10 operating system and R version 3.6. The execution of the binary classification methods took around 45 minutes, the exection of the feature ranking techniques took around 45 min and 30 s, while the execution of the biostatistics tests took around 5 seconds.
Discussion
In this section, we first discuss the results achieved by our binary classification for the survival prediction, and then we discuss the top predictive features detected by our computational intelligence approach, the top predictive features identified by our univariate statistical tests, and the top predictive features revealed by other studies.
Survival prediction
Our survival prediction results show that computational intelligence can effectively predict survival of patients from their clinical records, in few minutes and with small computational resources.
Our methods achieved good prediction results on all the confusion matrix rates (Survival prediction), and even outperformed the results obtained by the original dataset curators Santos et al. 27 which obtained a top performance score of ROC AUC = 0.700 through a neural network augmented sets approach. Our Random Forests classifier, in fact, obtained an average ROC AUC = 0.766 (Table 5). We believe that our improvement on the results, compared to the original study 27 is due mainly to the predictive power of Random Forests, 39 that often outperforms artificial neural networks and all the other machine learning techniques in health informatics binary classification tasks.19,74–77
Other studies have applied machine learning methods to predict survival and rank the clinical features on this HCC dataset (Dataset), but they included methodological mistakes that led to inflated and overoptimistic results. Ksikazek et al. 78 obtained inflated prediction scores due to several wrong machine learning practices: they split the dataset into a training set and a test set, used the test set for the hyper-parameter optimization of their Genetic Algorithm, and then applied their trained model on the same test set, generating high predictive results. The correct practice necessitates to splitting the dataset into three separate subsets: training set, validation set, and test set.34,36,43 The validation set should be employed for the hyper-parameter optimization, and the test set should be left untouched as a “held-out set” until the end, and used for the final classification made with trained optimized model. 79
In a parallel work, Sawhney et al. 80 committed a similar mistake: they decided to reduce the number of features of the dataset to predict survival, but they did it on the same subset they employed for testing their classification method. Again, the correct practice would have necessitated to splitting the dataset into three independent subsets: training set, feature reduction set, and test set. The “held-out” test set should have been used only at the end, after the training phase and the feature reduction phase. Additionally, the authors did not provide enough details on their feature reduction procedure.
Because of these malpractices, from a data analytics point of view, we did not compare our predictions performance with the results achieved by Ksikazek et al. 78 and Sawhney et al. 80 because the latter are biased and optimistic due to several data snooping issues (for example, some of the training data instances were employed also in the test set). 81
Clinical feature ranking obtained by our Random Forests approach
Regarding feature ranking, our machine learning approach identified clinical factors already known to be HCC predictive or prognostic factors in the gastroenterology community (Table 6).
Yu et al. 82 and Parikh and Sawant 83 for example, confirmed the predictive power of alkaline phosphatase (ALP) level for survival of patients having hepatocellular carcinoma. ALP is the top most predictive clinical factor found by our approach (Table 6). On the second position of our ranking we found alpha-fetoprotein (AFP), which was found to be strongly correlated to survival of patients having HCC by Tangkijvanich et al., 84 Johnson, 85 Johnson and Williams, 86 and Tyson et al., 87 in studies independent from each other. Finkelmeier et al. 88 confirmed the predictive importance of Hemoglobin level, which is on the third position of our ranking. The fourth position of our ranking lists the Albumin level, which has been confirmed to be related conditions of patients having hepatocellular carcinoma by Carr and Guerra 89 and Tanriverdi. 90
Regarding the Ferritin level found in the blood of patients, listed on fifth position of our ranking, the scientific literature contains several studies confirming this association.91–93 However, since the Ferritin feature has almost half of its values missing in the the original dataset (Dataset), we have to warn that the data imputation technique we used might have influenced this outcome.
The fact that our approach ranked factors already known to the gastroenterology community as most predictive of survival for patients having HCC confirms the effectiveness of our feature ranking approach. Interestingly, our approach listed ALP level, AFP level, Hemoglobin level, and Ferritin level in positions higher than other known HCC clinical factors.
The last five positions of our ranking, in fact, list Alcohol consumption, Cirrhosis, Sex, Radiological Hallmark, and Obesity as least predictive features for HCC patient survival. Even if the daily consumption of alcohol is known to be related to to hepatocellular carcinoma, 94 our analysis suggests it is unrelated to survival. Regarding Cirrhosis, even if often present in patients with hepatocellular carcinoma, 95 our ranking recommends it as non-predictive of survival. A study by Sangiovanni et al. 96 confirms the increased chances of survival for patients with HCC and cirrhosis. Our analysis suggests that the patient’s sex is not prognostic of survival, even if HCC is more common among men. Even if the hepatocellular carcinoma diagnosis is confirmed by the Radiological Hallmark, our study states that this aspect cannot say anything about possible survival or decease of the patient. Obesity is a known risk factor for HCC, 97 but our study suggests, that is, independent from the survival of the patient.
Clinical feature ranking obtained by our univariate statistical tests
Regarding biostastistics univariate tests, we noticed these techniques identified as most relevant clinical factors the same top eight features found by Random Forests (ALP, AFP, Hemoglobin, Albumin, Ferritin, PS, AST, Symptoms), plus other ones that were on lowest Random Forests ranks. Our ensemble learning technique, in fact, put Direct Bilirubin on 12th position, Ascites on 16th position, and Metastasis on 23rd position out of 49.
Platelet count, Age, and Total Bilirubin result being more relevant than Direct Bilirubin in our machine learning ranking, confirming the effectiveness of Random Forests in this task. Platelet count, in fact, is a known prognostic factor in hepatocellular carcinoma, 98 but it was undetected as relevant feature by the univariate statistical tests (Table 6). Age is another important factor: older patients have less chance to survive HCC, 99 but Age was unseen as a key factor by the univariate statistical tests (Table 6).
Ascites is a prognostic factor employed by the Okuda HCC staging definition 8 that was introduced in 1985 and by the Chinese University Prognostic Index 13 in 2002, but was excluded from more common staging systems such as the TNM Classification of Malignant Tumors.16,17
Metastasis was detected as a significant factor by the univariate statistical tests, but not by our Random Forests ranking.
HCC most predictive clinical features according to other studies
In the scientific literature, other papers claim alternative survival or mortality factors for HCC patients, which we compare with our top ranked features.
Cai et al. 100 listed tumor size and vascular invasion as the most relevant clinical features for survival of patients with hepatocellular carcinoma. The study of El-Fattah et al. 101 instead, stated that age, race, tumor size, AFP level, the American Joint Committee on Cancer (AJCC) stage, and the year of diagnosis were the most relevant factors for survival in the medical records of HCC-diagnosed patients. Falkson et al. 102 identified impaired performance status, male sex, older age, and disease symptoms (jaundice and reduced appetite) as factors most mortality-related. The research study of Vauthey et al. 103 identified cirrhosis and vascular invasion as clinical aspects more correlated to mortality.
Treatment for HCC, albumin level, and TNM stage were the most predictive survival features found by Kawaguchi et al. 104 in a recent article. Singal et al. 105 listed AFP and being a male as two relevant signs of potential survival from HCC. The article of Chaudhari et al. 106 instead, listed age, stage of disease, multiplicity, tumor thrombosis, lymphovascular invasion, nodal and distant metastases and completeness of resection as relevant factors for patients survival from HCC.
Two of these studies confirmed the relevance of AFP level,101,105 which we ranked
Two studies listed male sex as a top predictive factor,101,106 which we listed in the last positions of our ranking (Table 5): this discordance leaves room for further analysis about this aspect.
Conclusion
Hepatocellular carcinoma is a type of liver cancer that affects tens of millions of people worldwide (14 million in 2012 1 ) and kills approximately 800 thousand individuals worldwide every year.2,3 Predicting survival and detecting the most relevant clinical features for patient survival can be extremely useful to better understand this disease and its medical markers.
In this context, machine learning can provide methods to analyze clinical records in a few minutes and suggest the most relevant clinical factors for survival. In this study, we analyzed a public dataset of medical data of 165 patients recorded in Coimbra (Portugal), which contain 50 features for each patient. We first applied a data imputation and oversampling approach to take care of the missing values and of the dataset imbalance. We then employed several data mining methods to discriminate the survived patients from the deceased patients, outperforming the classification results obtained by the original dataset curators Santos et al. 27 Afterwards, we took advantage of our top performing method (Random Forests) to rank all the clinical features based on their relevance in predicting survival, discovering that the most important features resulted were alkaline phosphatase, alpha-fetoprotein, Hemoglobin, Albumin, and Ferritin levels.
We found scientific publications confirming the association between these features and hepatocellular carcinoma, which confirmed the soundness of our approach. We also compared our top features with other prognostic factor combinations we found in the medical literature, and we noticed some studies which endorsed the role of alpha-fetoprotein. Our analysis therefore suggests the inclusion of alkaline phosphatase and Hemoglobin in any future HCC prognostic indexes.
Even if other studies confirmed the relevance of ALT and AFP in hepatocellular carcinoma survival our study lists them as the two most important variables for the first time. This discovery can have impact on clinical practice, suggesting physicians and medical doctors to focus on these two clinical factors of the blood tests.
Our methods can result particularly useful if employed in small hospitals or clinics where medical imaging techninques for CT scan or MRI are unavailable.
As a limitation, we admit that employing a single dataset from a single hospital has been a drawback in our study: if we had an alternative dataset from another site as a validation cohort, we could have used it to confirm our findings. Our results might not generalize well to other clinical records. We searched online for public alternative datasets of patients having hepatocellular carcinoma, but unfortunately we found none.
Another limitation of this study has been the absence of survival time in the dataset. If this feature were present in the dataset, we could have framed our analysis to understand how long a patient would have survived, through methods such as a stratified logistic regression. 107
In the future, we plan to expand our analysis by making a comparison between the speed, the resources employed, and the cost of making the decision employed by our computational intelligence approach and the same elements employed by medical doctors in a hospital settings. Unfortunately we did not have this information to perform this comparison in this study, but we hope to obtain it for the future.
Regarding future developments, we also plan to apply our approach to hepatocellular carcinoma data derived from high throughput sequencing technologies, such as transcriptomics data. 108
We also aim at applying our approach to datasets of patients having other diseases such as neuroblastoma, 109 breast cancer, 20 amyotrophic lateral sclerosis, 110 and heart failure. 107
Supplemental Material
sj-pdf-1-jhi-10.1177_1460458220984205 – Supplemental material for Computational intelligence identifies alkaline phosphatase (ALP), alpha-fetoprotein (AFP), and hemoglobin levels as most predictive survival factors for hepatocellular carcinoma
Supplemental material, sj-pdf-1-jhi-10.1177_1460458220984205 for Computational intelligence identifies alkaline phosphatase (ALP), alpha-fetoprotein (AFP), and hemoglobin levels as most predictive survival factors for hepatocellular carcinoma by Davide Chicco and Luca Oneto in Health Informatics Journal
Footnotes
Abbreviations
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Data availability
The dataset used in this project is publicly available on (University of California Irvine Machine Learning Repository under its license at: https://archive.ics.uci.edu/ml/datasets/HCC+Survival). Our software code is publicly available under the GNU General Public License v3.0 at: (![]() )
)
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.