
Abstract
Background
Hepatitis D virus (HDV) represents the most severe form of human viral hepatitis, associated with rapid progression to cirrhosis and increased liver-related mortality. Globally, an estimated 9-19 million individuals are anti-HDV positive. To ensure early detetion, current guidelines recommend screening all HBsAg-positive individuals or, at a minimum, those with defined risk factors.
Methods
This expert consensus paper updates the current landscape of HDV management. Recommendations were derived from a structured expert panel discussion, incorporating recent evidence and clinical guideline developments, with a focus on screening, diagnosis, and antiviral therapy.
Results
The panel emphasized the importance of systematic HDV screening in HBsAg-positive individuals. Therapeutic strategies aim at sustained HDV-RNA suppression and, ideally, HBV surface antigens (HBsAg) loss. Bulevirtide was recommended as a long-term monotherapy. Pegylated interferon alpha (PEG-IFNα), if used, should be limited to 48 weeks and tailored based on viral response and tolerability. Combination therapy with bulevirtide and PEG-IFNα may be considered in selected cases.
Conclusion
This consensus provides updated recommendations for the screening, diagnosis, and treatment of HDV infection, highlighting the role of bulevirtide and individualized therapeutic approaches. As the treatment landscape continues to evolve, combination regimens and novel agents currently under investigation may offer additional options in the near future.
Introduction & methods
Hepatitis D Virus (HDV) is the most severe form of human viral hepatitis. A panel of 9 international experts evaluated the current landscape and issued recommendations based on guidelines, advancements in treatment options, and newly available literature.
Results & discussion
Natural history of HDV
Dr Mario Rizzetto first described the delta antigen 1977 as a subset of hepatitis B virus (HBV) infection with more severe liver disease.1,2 The hepatitis D virion comprises the hepatitis D RNA genome, hepatitis D antigen (HDAg), and a lipoprotein envelope with HBsAg proteins. 3 HDV requires HBV surface antigens (HBsAg) for cell entry and cellular RNA polymerases for replication.3,4
Infection with HDV occurs under 2 circumstances: coinfection and superinfection (Figure 1). Coinfection occurs when patients are exposed to HBV and HDV simultaneously. These patients are at risk of developing acute liver failure (acute viral hepatitis), characterized by acute inflammation of the hepatic parenchyma or injury to hepatocytes. 95% of patients with HBV/HDV coinfection have viral clearance of both HBV and HDV. Superinfection occurs when patients with established chronic HBV (defined by the presence of HBsAg in serum) are exposed to HDV.
4
Patients with HDV superinfections are those likely to develop chronic HDV infection.
4

Chronic HDV leads to severe viral hepatitis, with higher hepatocellular carcinoma (HCC) rates, faster cirrhosis progression, and higher mortality risk than chronic HBV monoinfection.4,5 Spontaneous clearance of chronic HDV infection has been reported, with some anti-HDV-positive patients testing negative for HDV-RNA in subsequent visits.6,7
Detectable HDV-RNA links to increased clinical events. 9 While the interaction between hepatitis B and D is not fully understood, it may involve reciprocal interference of the replication of the two viruses. HDV interferes with HBV via IFN-dependent (upregulation of IFN-stimulated genes) and independent mechanisms, lowering HBV viremia.3,10 HDV prevalence and outcomes in HIV/HBV coinfection remain underexplored (1.2–25%). Coinfection with HIV and persistent HBV replication increases liver risks, including decompensation, HCC, and death.3,11
Epidemiology of HDV
HDV prevalence, epidemiological, and clinical features vary geographically. HDV prevalence generally correlates with HBV prevalence (e.g., >20% in parts of the Amazon basin, 20–40% in parts of Africa, <1% in North America). 12 However, some countries or regions (e.g., parts of China) experience higher HBV prevalence (7.18%) and relatively low HDV prevalence overall (2.63% HDV seroprevalence among HBV infections) with factors including HIV-1 infection influencing regional prevalence and distribution. 13 Mongolia has the highest reported prevalence of HDV infection worldwide, affecting approximately 60% of HBsAg-positive individuals. 14 HBV vaccination protects against HDV, reducing prevalence of HDV in high-income countries where vaccination has been implemented recently. However, HDV resurgence occurs in high-income countries due to risky behaviors and immigration.3,15 HDV seroprevalence peaks in high-risk areas in Africa and Asia, Mongolia is a typical example of a high endemic area for both HBV and HDV.16–18 Up to 10.6% of chronic HBV patients could be coinfected with HDV. 3 In different epidemiological studies, HDV infections vary from 12 to 72 million people worldwide.19–21, The prevalence of heterogeneity impacts clinical interpretation of the data. Since epidemiological data across the world are highly variable and HDV testing is lacking in many regions, it is impossible to determine the exact prevalence of HDV infections globally, highlighting the need for more surveillance data.17,22
Of 8 known HDV genotypes, HDV-1 is distributed worldwide, whereas HDV-2 to HDV-8 are seen more regionally. Genotype 1 is the most prevalent among the isolates discovered in nearly all regions worldwide. Genotypes 2 and 4 primarily affect individuals in Japan and Taiwan, potentially linked to a less severe form of the disease. Genotype 3 is predominantly found in the Amazon region, where it is associated with notably severe outbreaks of the disease. Genotypes 5 through 8 have been identified in the blood samples of individuals of African descent.
21
Main Takeaways (1) Global estimates of HDV prevalence are unreliable or unavailable due to data heterogeneity and testing gaps. (2) More surveillance data at the country level are needed.
Screening, testing, and diagnosis of HDV
Current guidelines for screening for HDV.

ALT: alanine aminotransferase; AST: aspartate aminotransferase; DNA: deoxyribonucleic acid; HBV: hepatitis B virus; HCV: hepatitis C virus; HDV: hepatitis D virus; HIV: human immunodeficiency virus.
As shown in Figure 2, Persons Who Inject Drugs (PWIDs) are at the highest risk for developing HDV in high- to middle-income countries.
21
Moreover, there are dynamic trends in the incidence of HDV within specific high-risk groups. For instance, the prevalence is increasing among men who have sex with men, often linked to drug use during sexual encounters, referred to as “slamming.”
28
Populations at high-risk for HDV infection in middle- to high-income countries. HBV: hepatitis B virus; HCC: hepatocellular carcinoma; HCV: hepatitis C virus; HDV: hepatitis D virus.
Diagnosing HDV begins with screening for HDV antibodies (anti-HDV), using enzyme-linked immunosorbent assays (ELISA). Over 90% of acute HDV infections yield positive anti-HDV within 2 months.
29
However, a positive anti-HDV test doesn’t imply immunity.
30
According to the expert panelists, anti-HDV-IgG, which develops over several weeks after acute infection and persists with chronic HDV infection, is the preferred screening test over anti-HDV-IgM, which is primarily detectable in serum 2–4 weeks after initial exposure and rarely used in clinical practice, due to its availability and accuracy. Moreover, low titer anti-HDV-IgM is present in some chronically coinfected patients, which is associated with increased disease activity.
31
Anti-commercial ELISA assays have restricted availability, particularly in the US. Anti-HDV tests, including point-of-care (POC) tests, are working across all genotypes.
32
Main Takeaways (1) The first step in a diagnosis or screening pathway is total anti-HDV-IgG; in the context of acute hepatitis, anti-HDV-IgM can be requested. (2) Anti-HDV-IgM is primarily detectable in serum 2–4 weeks after initial exposure and is rarely used in clinical practice. (3) Anti-HDV-IgG develops several weeks after acute infection and persists with chronic HDV infection.
Patients with anti-HDV + should be tested for quantitative HDV-RNA to confirm active infection.3,7
Serum HDV-RNA can be detected using qualitative and quantitative reverse transcription-polymerase (RT-PCR) assays; quantitative is preferred. 29 HDV-RNA tests face limitations due to lack of standardization, varying characteristics, and limited availability. Many rely on in-house protocols without proper controls or genotype coverage, raising concerns of false negatives.7,33 Standardized HDV-RNA assays are needed for validation across laboratories. A recent review by Wedemeyer et al. addressed the challenges in achieving accurate pan-genotype HDV-RNA quantification, highlighted its critical role in drug development and patient monitoring, and summarized key features of available HDV-RNA assays. 34 Clinicians should always be aware of the limitations of PCR assays as the variability in sensitivity and the inconsistent documentation of negative results obtained from various laboratories pose challenges in the interpretation of HDV-RNA data.
An optimized assay has been suggested using consensus sequences for amplification and the World Health Organization (WHO) International Standards.29,35,36 The new commercial assays perform well in detecting and quantifying HDV load across genotypes. 36 While data on the real-world performance of commercial assays is sparse, notable examples include the RoboGene HDV RNA Quantification Kit 2.0, Diapro HDV Quantitation Real-Time PCR kit, and EurobioPlex HDV kit. 34
According to advisors, patients with HDV should undergo testing for HCV and HIV due to shared transmission routes. 30 Non-invasive fibrosis scores may be useful
Non-invasive fibrosis markers are valuable for assessment in HBV and HCV, but their role in HBV/HDV coinfection is so far underexplored, lacking defined cirrhosis criteria.37,38 Delta-4 fibrosis score (D4FS) for chronic HDV needs further validation for cirrhosis identification. 39 Transient elastography (TE, FibroScan) appears promising, particularly for cirrhosis exclusion, and unpublished data from AASLD 2024 suggests that a cutoff of <7 kPa is associated with reduced risk of progression.40,41 Two larger real-world studies have now provided promising data and initial suggestions for cut-off values to exclude cirrhosis and confirm severe fibrosis or cirrhosis.42,43 The results show a better diagnostic accuracy of TE in detecting cirrhosis compared to APRI, FIB-4, and D4FS. 43 Recent studies have also evaluated performance of 3 plasma-based tests for HDV-RNA quantification: Robogene 2.0 HDV-RNA Quantification Kit 2.0 (Roboscreen GmbH; LOD 6 IU/mL on 7500 Fast Real-Time PCR System [Applied Byosystem]), EurobioPlex HDV PCR quantitative (Eurobio Scientific, LOD 100 IU/m) on CFX 96TM real-time PCR detection system [Bio-Rad]), and AltoStar HDV RT-PCR RUO Kit 1.5 (Altona Diagnostics, estimated LOD <10 IU/mL) on the AltoStar®AM16. 44 Findings indicated variation in assay quantification over time, suggesting a need for additional research to further improve quantification and establish appropriate cut-offs. Overall, the amount of HDV data is still limited compared to HBV or HCV, so larger studies or pooled analyses are needed for confirmation.
Liver biopsies remain the gold standard for grading and staging. 38
Liver histology in patients with HDV characteristically features more intense inflammation than in HBV or HCV and can even mimic autoimmune hepatitis (AIH). Rare reports even indicate disease exacerbation with interferon alfa. Interestingly, some reports describing an improvement in AIH features with bulevirtide45,46 Fatty liver may also be present.40,47,48
Other testing/modalities are employed, but limitations restrict their use in clinical practice
Serum HDAg should not be tested (or looked for) as it is present most of the time in the form of immune complexes, and it escapes detection with commercial ELISA assays 30 The United States Centers for Disease Control and Prevention is offering HDV genotype testing using a high quality assay system. However, there are no guidelines at present that recommend varying approaches to management based upon HDV genotype, including recommendations on antiviral therapy. This is because there are no well documented differential responses of different HDV genotypes to antiviral treatments. Anti-HDV-IgM indicates ongoing infection/replication, but its clinical use is rare. In cases that HDV-RNA testing is not available, anti-HDV-IgM might be considered as an alternative surrogate test when HDV is suspected. 30
Simplifying HDV testing
Frequent clinic and lab visits complicate patient follow-up for diagnosis. 49 Point-of-care HDV-RNA and reflex testing, proven effective for other viruses, offer immediate results and avoid return visits. 49 Lempp et al. 32 measured anti-HDV with 94.6% sensitivity and 100% specificity using an HDV rapid test. Reflex testing includes automatic anti-HDV screening for all HBsAg-positive cases. 49 Double-reflex testing involves testing for HDV-RNA if anti-HDV is positive, as per the expert panel.50,51
Current guidelines
HDV screening rates among HBV-positive patients are universally low.23,52 In the Veterans Affairs Medical System, less than 8% of HBsAg-positive patients and only 19% meeting “high-risk” criteria were tested for HDV.
23
Similarly, a US Midwest study found just 12% of HBsAg-positive patients were screened for anti-HDV.
52
Screening rates are even lower in low-income and lower-middle-income countries.
18
The main takeaway box summarizes recommendations for screening. Main Takeaways (1) HBsAg-positive patients should be tested at least once with total anti-HDV. (2) Regardless of previous HDV testing, HBsAg-positive patients with worsening conditions (e.g., elevated ALT levels or hepatic synthetic dysfunction or ongoing risk factors) should be retested using anti-HDV. (3) Patients who test positive for anti-HDV should be immediately tested for quantitative HDV-RNA PCR using the WHO reference standard (reflex testing, if available); if qHDV-RNA is positive, patients should receive care for both HBV and HDV. (4) Family testing of household contacts or sexual partners of patients with HDV should occur for HBV, and if positive, for HDV.
Endpoints in clinical trials
Surrogate markers as clinical endpoints in HDV infection aren’t well defined. 53 While undetectable HDV-RNA in serum 6 months after the end of therapy is widely accepted as a marker of treatment efficacy, it is not an ideal clinical trial endpoint for chronic HDV due to the risk of late relapses, which have occurred 5–8 years after successful PEG-IFN therapy.53,54 This limitation is partly attributed to the lower sensitivity of HDV-RNA assays used in earlier studies, which may have failed to detect residual viral replication.34,55 More data are necessary to validate this endpoint, not only for its broader application but also for therapies such as bulevirtide, to ensure consistent and reliable assessment of treatment outcomes.
Primary, secondary, and exploratory endpoints for HBV/HDV coinfection. 53
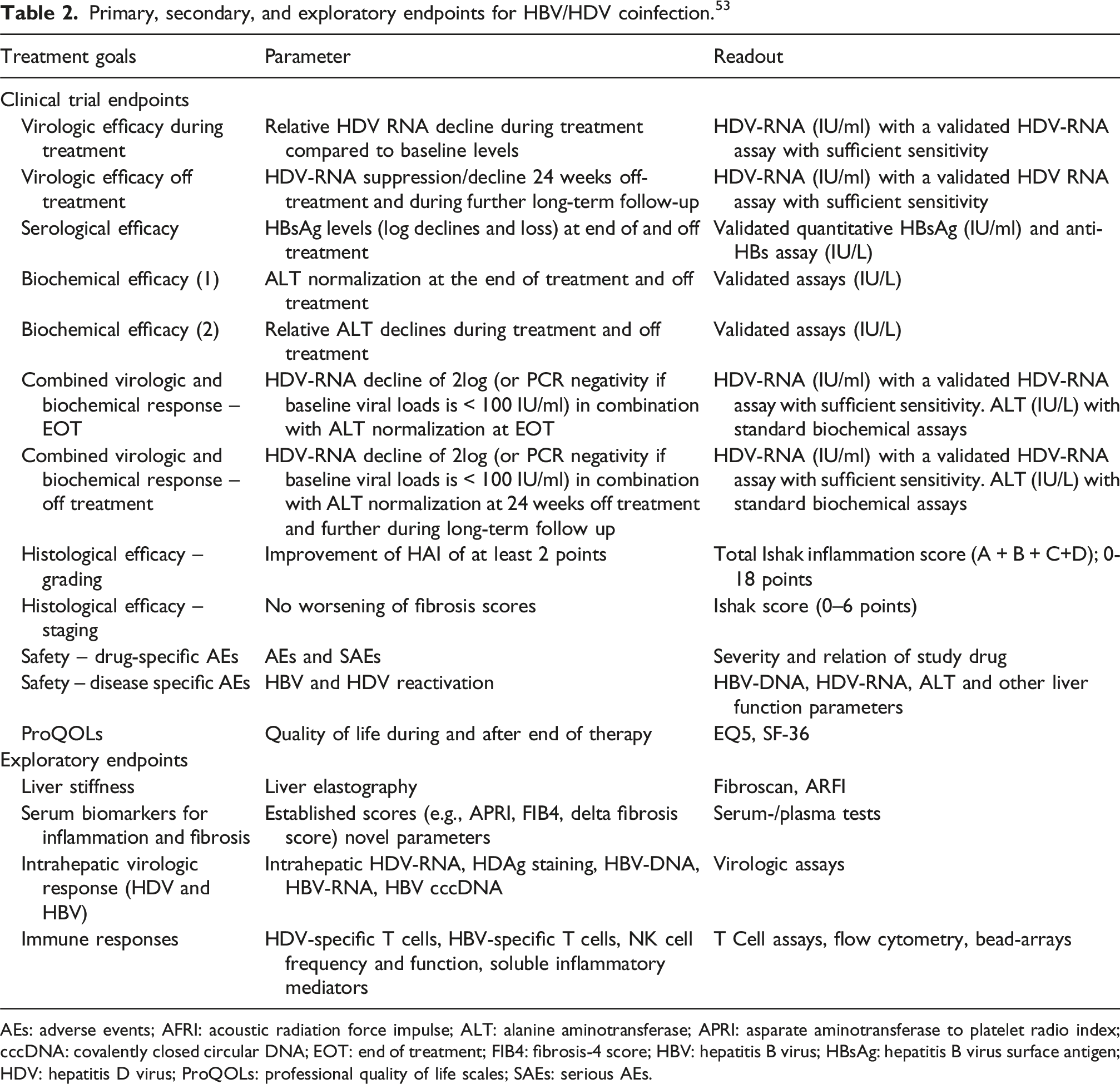
AEs: adverse events; AFRI: acoustic radiation force impulse; ALT: alanine aminotransferase; APRI: asparate aminotransferase to platelet radio index; cccDNA: covalently closed circular DNA; EOT: end of treatment; FIB4: fibrosis-4 score; HBV: hepatitis B virus; HBsAg: hepatitis B virus surface antigen; HDV: hepatitis D virus; ProQOLs: professional quality of life scales; SAEs: serious AEs.
Main Takeaways (1) Undetectable HDV-RNA in serum 6 months after end of therapy is considered an important endpoint, but it requires further validation as late relapses have occurred up to 5–8 years after successful therapy. (2) Success of finite treatment endpoints: • HBsAg loss (ideal). The timing of assessment varies according to treatment strategy. • HDV-RNA below the limit of quantification at 24 weeks off-treatment (there should be a commitment to long-term follow-up for a minimum of 5 years to assess durability of this endpoint with annual HDV-RNA quantitative testing). • ALT normalization (desired). The timing of assessment varies according to treatment strategy. • Improvement of necroinflammation and fibrosis. (3) Goals of long-term treatment: • ≥2 log reduction in HDV-RNA (but still detectable) combined with normalization of ALT level at 48 weeks on-treatment and during the follow-up on-treatment. • Undetectable HDV-RNA and normal ALT at the EOT in case of finite therapy. • The optimal duration of maintenance therapy is currently unknown.
Treatments
HDV treatment guidelines.

AASLD, American Association for the Study of Liver Diseases; APASL, Asian Pacific Association for the Study of the Liver; BLV, Bulevirtide; CHD, chronic hepatitis D; DGVS, Deutsche Gesellschaft für Gastroenterologie Verdauungs-und Stoffwechselkrankheiten; EASL, European Association for the Study of the Liver; HBIG, hepatitis B immune globulin; HBsAg, hepatitis B surface antigen; HBV-DNA, hepatitis B virus deoxyribonucleic acid; HDV, hepatitis D virus; IU/mL, international units per milliliter; NAs, nucleoside/nucleotide analogues; PEG-IFNα, pegylated interferon alpha; TAF, tenofovir alafenamide; TDF, tenofovir disoproxil fumarate; μg, microgram.
PegIFNα
Studies on IFNα include 2 randomized controlled trials and numerous uncontrolled trials, with prospective and retrospective design. In a systematic review of 15 studies, 17–47% of pegIFNα-treated patients showed virologic response at 24 weeks post-treatment. 59 However, over 50% of those may experience relapse up to 10 years post-treatment as shown by the Hep-Net-International Delta Hepatitis Intervention I (HIDIT I) study.54,60 PCR assay sensitivity for HDV-RNA detection has changed from >1000 IU/ml to 6 IU/ml over time, impacting result comparability of these trials. Upon re-testing samples with a more sensitive assay, 31% of previously negative samples were found positive, revealing potential misclassification due to suboptimal sensitivity, 61 which helps to explain the late relapse. Despite HDV-RNA recurrence, the extended HIDIT-I trial revealed better outcomes for patients achieving undetectable HDV-RNA at 6 months during post-treatment follow-up. 54
Extending IFNα treatment to 2 years in most studies doesn’t appear to increase virologic response rates.62–64 IFNα has been also employed in CHD patients with compensated cirrhosis. 24 Recognizing the heterogeneity of HDV and that the PegIFNα mechanism of action is not well understood, additional research is needed to understand factors impacting response.
Bulevirtide (BLV)
Bulevirtide (BLV) has been used as an antiviral drug since its approval by the European Medicines Agency (EMA) in July 2020 (full approval in July 2023) and is therefore a relatively new addition to HDV therapy and treatment.
Based on data from Phase 2 and Phase 3 studies, 65 BLV is administered subcutaneously at a dose of 2 mg/day for the treatment of compensated CHD. The phase 3 study showed that 48 weeks of BLV therapy decreased HDV replication, normalized ALT levels in ∼50% of patients, and improved quality of life.66,67 BLV was well tolerated, with frequent reports of increased bile acids due to BLV’s mechanism of action (binding to sodium taurocholate co-transporting polypeptide) and injection site reactions due to subcutaneous injections. 66 Real-world studies in chronic HDV and compensated cirrhosis confirmed these promising results.45,68,69 A recent follow-up of the 48-week phase 3 study presented at EASL in June 2023 found BLV to be a safe and well-tolerated monotherapy for CHD up to week 96, with further improvement in virologic and biochemical response observed with longer treatment duration. 73 However, the duration of BLV treatment required to achieve complete virologic control without relapse remains uncertain. Recent real-world data confirm that prolonged suppressive therapy with BLV monotherapy has the potential to reduce viral replication in the long term to such an extent that it may be possible to stop therapy even without HBsAg loss. 70 Randomized clinical studies on the use of BLV in decompensated liver cirrhosis are currently not available. However, patients with decompensated cirrhosis have already been treated in real-world studies 71 and there were no safety concerns, so that therapy is recommended in individual cases after risk/benefit assessment. 58
Combination of BLV with PegIFNα
IFNα′s immune modulation, antiviral effects on HBV and HDV, and capacity to hinder HDV spread support its combination with BLV, targeting a lasting virological response.24,72 In the ongoing MYR204 study, patients were randomized to receive 2 or 10 mg BLV with pegIFNα for 48 weeks, then BLV for another 48 weeks, or 10 mg BLV monotherapy for 96 weeks. The primary endpoint was HDV-RNA undetectable at week 24 after EOT, and the secondary endpoints were undetectable HDV-RNA at EOT and change from baseline in liver stiffness at week 24 after EOT. Additional endpoints included ALT normalization and composite response.
The efficacy and safety of BLV in combination with pegIFNα results were presented recently at AASLD from the phase 2b, open-label, randomized, multicenter study (MYR204). The data demonstrated that in combination with PegIFNα, BLV 10 mg demonstrated the highest rates of HDV-RNA undetectability, ALT normalization, and composite response, surpassing both BLV 10 mg monotherapy and PegIFNα monotherapy (primary endpoint). 73 Additionally, BLV 2 mg in combination with PegIFNα outperformed BLV 10 mg monotherapy in terms of HDV-RNA undetectability and composite response. Specifically, the combination therapies showed improvements in liver stiffness observed across the BLV-containing treatment groups. HBsAg loss was infrequent but observed in the combination groups. BLV combined with PegIFNα was safe and well-tolerated as the safety profile observed with BLV (10 mg or 2 mg) in combination was similar to pegIFNα alone. Moreover, few Grade 3 treatment-emergent adverse events (TEAEs) and no serious adverse events (SAEs) related to BLV were reported. 73 The study also measured the total serum bile acids and found dose-dependent asymptomatic elevations in bile acids in the BLV groups, attributed to the BLV mechanism of action. However, the increase in the bile only occurred early and the mean levels were stable with continued treatment and even reversible upon treatment completion.
De novo combination therapy shows promise, but studies have had small patient numbers and unexplained differences between dose groups. In a small cohort from Vienna, patients who did not achieve further decreases in HDV-RNA after 24 weeks were offered PEG-IFN as an add-on therapy in a response-guided manner. 74 The optimal duration for BLV treatment, alone or in combination with PEG-IFN, remains undetermined. More studies are needed on combination therapy for CHD to optimize treatment schedules before proposing it as first-line therapy aiming at curing HDV infection. Currently, BLV plus pegIFNα should be considered for compliant patients within clinical protocols. 24
Considerations for anti-HBV therapies
Developing HDV antiviral agents is challenging because it uses host enzymes for replication and can replicate independently of HBV. These viral properties make antiviral agents like nucleos(t)ide analogues (NAs) ineffective. Studies showed NAs like famciclovir, lamivudine, entecavir, and adefovir lacked efficacy against HDV either as monotherapy or in combination with IFNα.75–78
However, tenofovir disoproxil fumarate (TDF) showed improvement (HDV-RNA and fibrosis) in HIV-coinfected HDV patients, 79 possibly by restoring the immune system in the HIV-infected cohort or inducing interferon lambda. 80
In summary, there is no evidence of benefit for NAs therapy in chronic HDV.81,82 Nevertheless, the treatment principles for HBV monoinfections also apply to coinfected HBV/HDV patients, i.e., NA therapy is indicated if HBV DNA is ≥ 2000 IU/mL or if HBV DNA is positive and advanced fibrosis or cirrhosis is present.24,58
Liver transplantation
Liver transplantation stands as a viable choice for individuals with end-stage liver diseases. Post-transplant, HBsAg and HDV-RNA clearance is pursued through hepatitis B immune globulin (HBIg) and NAs. 83 HDV-RNA decreases after liver transplantation,84,85 while anti-HDV-IgG and HDV total antibodies persist for years, according to experts. Immunohistochemistry analysis of liver biopsies can detect HDAg up to 590 days post-transplant in HDV-induced liver disease, without detectable ongoing HBV replication. This presence of HDAg in post-transplant livers is not associated with detectable HDV-RNA in the patient’s bloodstream, indicating that HDV in the liver may remain dormant but could potentially reactivate if the patient experiences a superinfection with HBV. This underscores the rationale for considering extended prophylaxis with HBIg and with high genetic barrier NA. 84
If HDV is present in liver cells it may reproduce, but it is unable to spread in the absence of HBV particles, as HDV lacks HBs proteins expression. While reinfection can occur post-transplant, it is a rare event due to continuous HBIg and NA prophylaxis recommended by major guidelines24,86–88
New therapies against HDV 89
(i) Preventing Assembly of Mature HDV Particles: Lonafarnib inhibits HDAg prenylation of large delta antigen (L-HDAg), crucial for HBsAg interaction.
17
Phase 2 studies tested different lonafarnib dosing regimens, alone or with pegIFNα or ritonavir. In LOWR HDV-1,
90
higher lonafarnib doses lowered HDV viral load, yet triggered more gastrointestinal (GI) events. Adding pegIFNα or ritonavir improved antiviral responses and reduced events compared to lonafarnib alone.
90
In LOWR-2 study, combining low-dose lonafarnib, ritonavir, and pegIFNα achieved maximum efficacy; again, high-dose lonafarnib resulted in more GI events.
56
The ongoing D-LIVR phase 3 study investigates lonafarnib (LNF) + ritonavir (RTV) ± pegIFNα in chronic HDV patients on anti-HBV nucleos(t)ide maintenance therapy.
91
According to the 48-week data, both LNF-based regimens, (LNF + RTV, 12.6%) and (LNF + RTV + pegIFNα 24.2%) demonstrated success in meeting the primary efficacy endpoint (the composite virologic and biochemical response rate at end-of-treatment in patients who receive LNF 50 mg/RTV 100 mg BID), outperforming the placebo (2.6%). Furthermore, it is noteworthy that the combination arm showed statistically significant histological improvement.
92
The 72-week follow-up data, presented in the 2023 EASL meeting, revealed that the combination was still showing consistent endpoint response and the treatment was well tolerated in both arms. (ii) Preventing Export of HDV Particles: Nucleic acid polymers (NAPs) hinder HBsAg secretion, which in turn can result in blocking HDV secretion from infected cells.
93
Phase 2 studies tested NAPs with pegIFNα via infusion in a few CHB/CHD cases, showing off-treatment HBsAg clearance after 63 weeks in several patients.
94
Off-treatment ALT flares are a potential issue. Several case reports from the Compassionate Use Program show promising results in patients who have failed PEG-IFN and/or BLV therapy.
95
Further research is required. (iii) Use of PegIFN-lambda: PegIFNλ is expected to have similar efficacy to pegIFNα but fewer side effects.
96
A phase 2 study has reported treatment of chronic HDV with PegIFNλ for 48 weeks, which led to a durable virologic response and was associated with a favorable side effect profile.
97
The phase 3 study investigating the use of 48-week treatment with pegIFNλ has been stopped due to safety concerns because some patients experienced hepatic decompensation.
98
(iv) Monoclonal antibodies against HBsAg with neutralizing activity, RNA interfering drugs (ASO, siRNA) entered the clinical evaluation, but additional research is needed to validate their use in larger trials and real-world clinical settings.
99
The Supplemental Table includes additional detail regarding emerging HDV therapies. Main Takeaways (1) Patients with mild disease don’t require immediate therapy and may await new treatments’ availability. (2) PegIFNα for 48 weeks is an option for finite therapy. Treatment beyond 48 weeks generally doesn’t increase overall response rates but some patients benefit from prolonged pegIFNα treatment beyond 1 year.100,101 Long-term treatment, 5 years follow-up, indicated that liver-related clinical events were uncommon and less likely to occur in patients who experienced virological responses to PEG-IFNα treatment.
102
Reduced dosage with prolonged treatment has been described in case reposts and case series. (3) Predicting who will respond to pegIFNα is crucial, as new treatments will not become available soon, particularly in HDV endemic countries. (4) BLV has received approval in Europe by the EMA and represents a breakthrough in the treatment of CHD. However, it is not yet available in other parts of the world, and the treatment duration has not yet been determined (5) Defining which patients benefit from the combination of pegIFNα plus BLV (de novo, add-on, or sequential) is important. Patients with inadequate response to BLV may benefit from add-on pegIFNα treatment. (6) HBV NAs are recommended when HBV-DNA ≥2000 IU/mL or in cirrhotic patients with advanced fibrosis or cirrhosis with any detectable HBV-DNA.
Post-treatment and monitoring
Follow-up after HDV treatment varies depending on the treatment regimen and outcomes. Patients treated with pegIFNα with detectable HDV-RNA at EOT may experience subsequent clearance within 24 weeks, 75 and on the other hand, late relapses have been reported. 55 Therefore, the advisors recommended a long follow-up period, even in noncirrhotic cases, considering re-treatment or a novel therapy if viremic. Re-treatment urgency might be less with normal ALT; however, clinicians need more data on response maintenance and long-term outcomes, such as fibrosis progression and clinical complications, according to the panel discussion. Noninvasive fibrosis monitoring, such as transient elastography, helps for ongoing management. 40 Patients with CHD failing treatment response should be monitored at regular intervals for progressive fibrosis which, if found, can lead to efforts to employ novel therapies. 103
The increased risk of HCC in patients with HDV appears to be related in part to the incremental risk of cirrhosis. 104 The unclear HCC risk in non-cirrhotic HDV patients might prompt broader screening and surveillance. Consistently, guidelines suggest HCC screening and surveillance for all HDV patients. Additionally, incremental HCC risk exists in HIV/HBV/HDV co-infected patients, warranting screening. 5
Conclusions
Progress has been achieved in addressing HDV, marked by approval and improved access to bulevirtide and updated EASL guidelines in 2023. Further investment and information are crucial to enhance awareness, screening, and the long-term efficacy and safety of therapies. Following the recommendations of clinical experts can lead to improved detection and early intervention of patients with HDV.
Supplemental Material
Supplemental Material - Best practices for screening, testing, diagnosing, and treating patients with hepatitis D (delta) virus based on global expert review and recent guidelines
Supplemental Material for Best practices for screening, testing, diagnosing, and treating patients with hepatitis D (delta) virus based on global expert review and recent guidelines by Markus Cornberg, Fabien Zoulim, Robert Gish, Ira M. Jacobson, Tatyana Kushner, Pietro Lampertico, Mario Rizzetto, Cihan Yurdaydin and Michael Manns in Antiviral Therapy
Ethical considerations
Due to the nature of the review article, ethical approval was not required.
Footnotes
Acknowledgements
The authors would like to thank Ellen Whipple, The Write Source, and Guanine Medical Inc. for medical writing support and Kelly Schrank for editorial support.
Author contributions
All authors contributed equally to the writing of the manuscript. Prof Dr Manns provided author coordination and editorial support.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support was provided by Gilead Sciences.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MC: Abbvie – lecturer, consulting, travel support; Gilead Science – lecturer, consulting; Falk Foundation – lecturer; Merck (MSD) – consulting, lecturer; AiCuris – consulting; GlaxoSmithKline (GSK) – lecturer, consulting; Novartis – consulting; Roche – consulting; AstraZeneca – consulting. FZ: lecturer and/or consulting: Aligos, Assembly, AusperBio, Blue Jay, Gilead, GlaxoSmithKline (GSK), nChroma, Precision Biosciences, Vir Bio; research grants: Assembly, AusperBio, Blue Jay, ImCheck. RGG – Grants: Gilead, Consultant/Advisor: Abacus, Abbott, AbbVie, Albireo, Aligos, Altimunne, Antios, Arrowhead, AstraZeneca, Audentes Therapeutics, Corcept, Dynavax, Effectus, Eiger, Eisai, Enyo, Genentech, Genlantis, Gerson Lehrman Group, Gilead Sciences, GlaxoSmithKline, Helios, HepaTX, HepQuant, Intercept, Janssen, JBS Science, Kinnate Bio, Merck, Precision BioSciences, Pfizer, Seres Therapeutics, Topography Health, Tune Therapeutics, Venatorx, Virion, Fibronostics, Fujifilm/Wako, Perspectum, Quest, Sonic Incytes; Scientific or Clinical Advisory Boards: AbbVie, Dynavax, Enyo, Genentech, Genlantis, Gilead, Helios, HepaTX, HepQuant, Intercept, Janssen, Merck, Pfizer, Prodigy; Clinical Trials Alliance: Topography Health; Chair Clinical Advisory Board: Prodigy; Data Safety Monitoring Board: Arrowhead, CymaBay Therapeutics, Durect, Kezar Life Sciences, Sagimet, Takeda; Speaker: AbbVie, AstraZeneca, BMS, Diasorin, Eisai, Genentech, Gilead Sciences Inc., Intercept, Mallinckrodt, VBI Vaccines; Minor stock shareholder: RiboSciences, CoCrystal. IJ – Consulting: Aligos, AusperBio, Barinthus, Gilead, Moderna, Roche, Vir Bio; Data Monitoring Committee: GSK, Precision Biosciences; Research funding: Assembly, Ausperbio, Bluejay, GSK, Vir Bio. TK: Gilead, Abbvie, Mirum, Ipsen, Madrigal, Eiger, GSK – Advisory; Gilead, Mirium, Abbvie – Research support. PL: Advisory Board/Speaker Bureau for Roche Pharma/Diagnostics, Gilead Sciences, Gsk, Abbvie, Janssen, Myr, Eiger, Antios, Aligos, Vir, Grifols, Altona, Roboscreen. MR: Lecturer – Gilead. CY: lecturer and/or consulting: Gilead, Roche, Eiger. MM: Abbvie – lecturer, travel support; Assembly Bio – consulting; Bristol Myers Squibb – consulting, lecturer, travel support; Falk Pharma – consulting, lecturer, travel support, monitoring board/data analysis; Gilead – consulting, lecturer, travel support, monitoring board/data analysis; Merck – principal investigator, grant, consulting, lecturer, travel support, monitoring board/data analysis; GSK – consulting.
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.