
Abstract
Background
Human cytomegalovirus (HCMV) is involved in complications on immunocompromised patients. Current therapeutics are associated with several drawbacks, such as nephrotoxicity.
Purpose:
As HCMV infection affects inflammation pathways, especially prostaglandin E2 (PGE2) production via cyclooxygenase 2 enzyme (COX-2), we designed 2'-hydroxychalcone compounds to inhibit human cytomegalovirus.
Study design
We first selected the most efficient new synthetic chalcones for their effect against COX-2-catalyzed PGE2.
Study sample
Among the selected compounds, we assessed the antiviral efficacy against different HCMV strains, such as the laboratory strain AD169 and clinical strains (naïve or multi-resistant to conventional drugs) and toxicity on human cells.
Results
The most efficient and less toxic compound (chalcone 7) was tested against HCMV in combination with other antiviral molecules: artesunate (ART), baicalein (BAI), maribavir (MBV), ganciclovir (GCV), and quercetin (QUER) using Compusyn software. Association of chalcone 7 with MBV and BAI is synergistic, antagonistic with QUER, and additive with GCV and ART.
Conclusion
These results provide a promising search path for potential bitherapies against HCMV.
Highlights
• We designed 2’-hydroxychalcone compounds to inhibit human cytomegalovirus (HCMV). • Among those compounds, chalcone 7 shown in vitro antiviral efficacy against laboratory and clinical HCMV strains and low cell toxicity. • We associated chalcone 7 with various antiviral molecules. • We observed a synergistic effect when chalcone 7 is combined to maribavir or baicalein.
Introduction
Inflammation is a complex physiological response to infection or injury, involving activation of various immune cells. Thus, macrophages participate in this biological process by producing various inflammatory mediators such as cytokines, prostaglandins (PGs), and nitric oxide. 1 Key regulatory enzymes are cyclooxygenases (COXs) which mediate the conversion of arachidonic acid, released from phospholipid membranes, to PGs. Two isoforms of COX, COX-1 and COX-2, have been identified. 2 COX-1 is a constitutive enzyme expressed in different tissues and acts on maintaining the physiological level of PGs. COX-2 is inducible by different agents like proinflammatory cytokines, hormones, and tumor promoters. Thus, COX-2 appears to be the dominant source of PGs formation at the inflammatory site. 3
Human cytomegalovirus (HCMV) is an opportunistic virus involved in severe complications in immunocompromised hosts like transplant recipients. The virus interferes with different pathways of oncomodulation to increase its own replication.4,5 One of these oncomodulatory pathways is the production of PGE2 that promotes inflammatory conditions. It was demonstrated that the amount of COX-2 increases when cells are infected with HCMV, 6 and that COX-2 is needed for viral infection since PGE2 performs an important function to support viral replication.7,8 Furthermore, non-selective COX inhibitors, such as aspirin and indomethacin, inhibit HCMV infection in vitro and block cell-to-cell spread.8–10
Aspirin and indomethacin belong to nonsteroidal anti-inflammatory drugs (NSAIDs), which are worldwide used for their anti-inflammatory and analgesic effects, but are associated to serious adverse effects like gastric bleeding or perforation and hepatotoxicity 11 due to the COX-1 inhibition. Thus, COX-2 selective inhibitors were developed to reduce these adverse effects. Structure-activity relationship studies revealed the key determinants for COX-2 inhibitory activity such as a 1,2-diaryl heterocyclic or carbocyclic structure and a sulfonamide or a methylsulfonyl substituent at para position of one of the aromatic rings. 12 However, some of them were found to be responsible for severe cardiac side effects. 13 The market removal of rofecoxib and some other COX-2 inhibitors clearly encourages the exploration and evaluation of alternative templates with COX-2 inhibitory activity and enhanced safety profile. Currently, many compounds identified from plants, such as polyphenols, were reported to inhibit COX-2 expression with little side effects. 14
Chalcones refer to a subcategory of the flavonoid family that comprises a wide variety of polyphenolic natural products. Chalcones are open-chain flavonoids in which the two aromatic rings are bridged by an enone linkage. They are abundant in edible plants and are considered as the main precursors for the biosynthesis of other flavonoids. 15 The biological properties of chalcones are equally wide ranging. The pharmacological activities associated with them and their related derivatives include anti-mitotic, 16 anti-bacterial, 17 anti-malarial, 18 anti-oxidant, anti-tumor, and anti-inflammatory properties. 19 With regard to the anti-inflammatory activity, some reports indicate that derivatives of chalcone and those of 2’-hydroxychalcone are able to inhibit COX-2 as well as this enzyme catalyzed PGE2 production.20–22 Moreover, flavonoids, including chalcones, have interesting anti-viral properties against herpesviruses including CMV.23–25
As part of our efforts directed toward the development of novel flavonoid derivatives as potent HCMV growth inhibitors, we report here the synthesis of new 2’-hydroxychalcone compounds. Considering the relationship between PGE2 production and HCMV infection, the chalcone derivatives were designed to present PGE2 synthesis inhibitory properties. Thus, the sulfonamide moiety, which is a COX-2 pharmacophore, and other substituents (chlorine, fluorine, and methyl group), that seem to improve the expected inhibitory activity, 26 were introduced onto the 2’-hydroxychalcone skeleton. Therefore, chalcone derivatives were first evaluated for their inhibitory effects against COX-2-catalyzed PGE2 production from lipopolysaccharide (LPS)-induced RAW 264.7 cells. Then, the most active compounds were selected for further evaluation of their antiviral activity against HCMV. Combinations of one of these chalcone derivatives with other antiviral compounds were also tested, as the association of valganciclovir and the specific COX-2 inhibitor celecoxib prevented HCMV replication in vitro. 27
Methods
Compounds
Compounds are shown in Figure 1(a) and numbered from 1 to 12. The 2’-hydroxychalcone ( (a) Chemical structure of 2’-hydroxychalcone. (b) Synthesis of 2’-hydroxychalcone derivatives 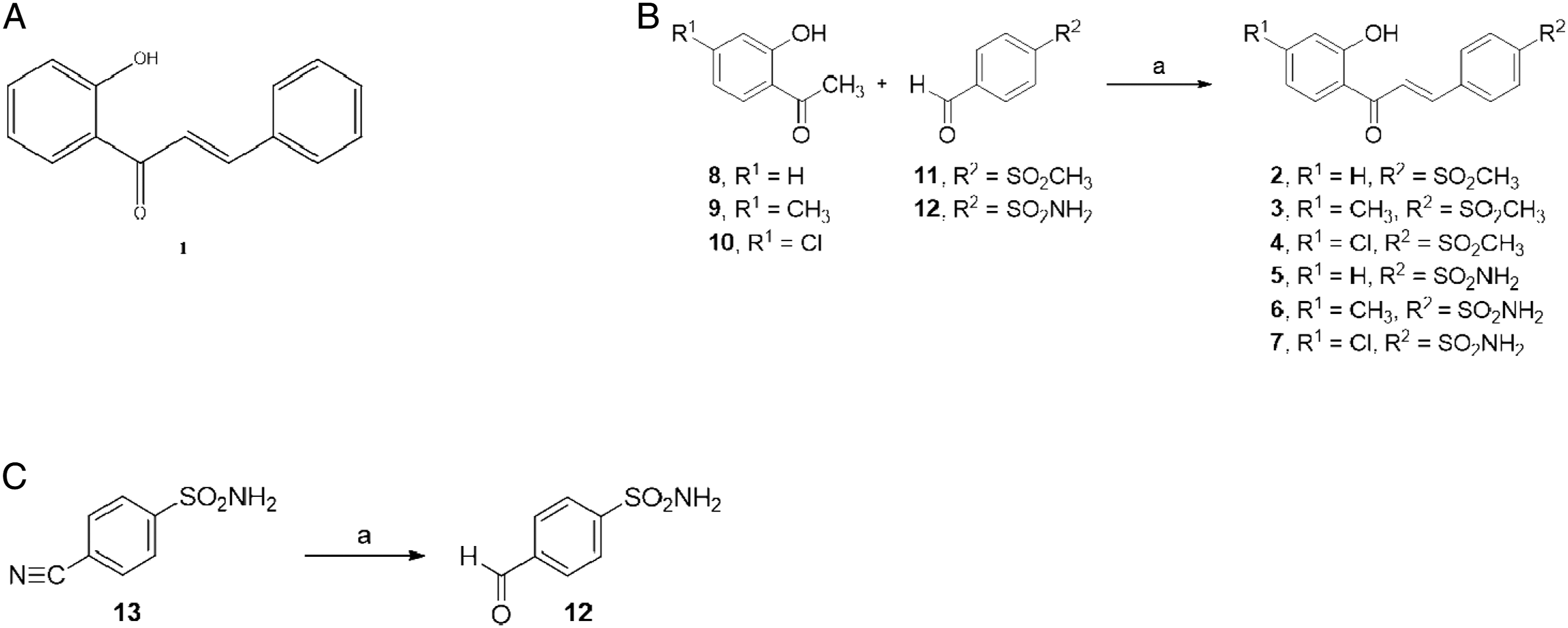
The preparation of 2’-hydroxychalcone derivatives (
2’-hydroxychalcone
Cell culture and viruses
Raw 264.7 murine macrophages (ATCC TIB 71; American Type Culture Collection, Rockville, MD, USA) were maintained in DMEM supplemented with 10% (v/v) heat-inactivated fetal calf serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. The cells were grown in a humidified incubator at 37°C and 5% CO2.
Human embryonic lung (HEL) fibroblasts (MRC-5) (bioMérieux, Lyon, France) between passages 25 and 35 were cultured at 37°C in a 5% CO2 atmosphere, in modified Eagle’s medium GlutamaX (Eurobio, Les Ulis, France) supplemented with 10% fetal bovine serum (FBS), penicillin (1000 IU/ml), and gentamicin (0.01 mg/mL).
For antiviral assays, we used a fluorescent laboratory strain AD169-GFP (gift from Eva Borst and Martin Messerle, University of Hannover, Germany) and three clinical isolates: TROA07, a naïve isolate from congenitally infected newborn, and two drug-resistant isolates, CHAJ06, a multidrug resistant strain, with an UL97 C592G and UL54 N408K and G841A substitutions, and JAMP01, a GCV-resistant strain, with an UL97 A595S substitution.
Inhibition of PGE2 production in RAW 264.7 murine macrophages
Raw 264.7 murine macrophages were seeded at 2.105 cells/well during 24 h, then 10 μM of each synthesized derivatives was added. After 2 h, LPS (10 ng/mL) was added in the culture medium during 24 h. The PGE2 levels were quantified in culture media supernatants from treated and control cells by enzyme immunoassay using an EIA Kit (Cayman Chemical) as previously described. 30 The inhibition percentages of these compounds on PGE2 production were calculated, considering that the inhibition percentage of NS-398, a well-known COX-2 selective inhibitor used as reference, was 100%. The results were expressed by the standard deviation average of ±3 independent experiments.
Anti-proliferative assay in Raw 264.7 murine macrophages
Anti-proliferative effect of molecules was determined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described. 31 Raw 264.7 murine macrophages were seeded in 96-well culture plates and grown for 24 h, in culture medium prior to exposure or not to molecules. MTT assay was performed 24 h after treatment, and cell viability was expressed in percentage of each condition of treatment by normalizing to untreated cells. Experiments were performed in triplicate.
ELISA assay for evaluation of PGE2 production
The different compounds were added at a concentration of 4-fold EC50, on MRC-5 48-well plate, with or without AD169-GFP. After 5 days of incubation, the supernatants were harvested and analyzed using a PGE2 ELISA kit (ab133021, Abcam, USA) according to the manufacturer’s protocol.
Briefly, standards, controls, and supernatants were added to 96-well plate precoated with a mouse IgG antibody, along with an alkaline phosphatase (AP) conjugated-PGE2 antibody. After incubation, the excess reagents were washed away; pNpp substrate was added and catalyzed by AP to produce a yellow color. The intensity of the yellow coloration was inversely proportional to the amount of PGE2 captured in the plate.
Plates were read on a PerkinElmer EnVisionTM plate reader, using an excitation wavelength of 405 nm. PGE2 levels were expressed as ng/mg of protein.
Cytotoxicity assay in MRC-5
Toxicity of compounds and associations was measured using the CytoTox96 Non-Radioactive cytotoxicity assay (Promega, Charbonnières, France), in confluent or 10% confluent MRC-5 in 96-well plates. This assay determines the lactate dehydrogenase (LDH) activity in the residual cells after incubation with the tested drug. Five days post-treatment, LDH activity was determined for each drug concentration by measuring the absorbance at 490 nm as compared to control wells (same compound concentration without cells). The 50% cell cytotoxicity (CC50) and 90% cell cytotoxicity (CC90) were determined graphically as the concentration of molecule causing 50% and 90% cell-death, respectively.
Antiviral plaque reduction assay
A plaque reduction assay was used to measure the concentration of drug required to reduce the number of plaques by 50% (50% effective concentration, EC50) as compared to controls without drug. Antiviral assays were conducted as previously described with some modifications. 24 Briefly, a 48-well plate of MRC-5 confluent cells was infected with AD169-GFP-infected cells or cell-free virion stock from isolates (prepared as described previously), 24 at a multiplicity of infection (MOI) of 0.01 to reach 25 to 50 plaques per well. After 3 h incubation, the medium was removed and cells were incubated with a range of drug concentrations for 5 days. Foci of infected cells were visualized on optical inverted microscope (or fluorescent microscope for AD169-GFP) and counted; EC50 was determined graphically from the dose-response analysis. Sensitivity index (SI) was calculated for clinical isolates as the ratio of their EC50 for an antiviral on the EC50 of the wild-type strain (SI 50 > 3 correlates to a resistant strain).
Western blot analysis
To investigate the viral replication step inhibited by these new anti COX-2, western blot analysis was performed. The different compounds and indomethacin were added, respectively, to 25-cm2 flask of AD169-GFP-infected MRC-5 (MOI 0.01) at a concentration two-fold higher than their EC50. After a 5 days incubation period, cells were washed with cold PBS, and 500 μL of RIPA lysis buffer (150 mM sodium chloride, 1.0% NP-40, 50 mM Tris, pH 8.0) supplemented with anti-proteases (1/1000, Sigma, Ilkirch, France) was added. After 5 minutes at 4°C, the cell-lysate was harvested and centrifuged at 10 000 g at 4° C. Supernatant was stored at −20°C. Samples were mixed with equal volume of Laemmli buffer, heated 10 min to 94°C with 1% DTT and 1% β-mercaptoethanol, and 20 μL of mix was loaded onto a 6% sodium dodecyl sulfate polyacrylamide gel. After electrophoresis, proteins were transferred onto a blot membrane using a Bio-Rad transfer system. Viral proteins were detected using mouse monoclonal antibody anti-HCMV immediate early IE1-72 and IE2-86 proteins at a 1/40 dilution in PBS (E13, bio Mérieux, Meylan, France) and revealed with the Clarity™ Western ECL Substrate (Bio-Rad, France), under UV light, following provider’s instructions.
Synergy assay
MRC-5 cells were infected in the same conditions as for a plaque reduction assay. COX inhibitors were associated with other antiviral compounds: artesunate (ART), maribavir (MBV), quercetin (QUER), baicalein (BAI), or GCV. Associations were tested using 2-fold EC50, EC50, ½ EC50, and ¼ EC50, for each compound. Data were analyzed with Compusyn© software, 32 and the combination index (CI) was calculated for each association in constant ratio of compounds. Values of CI determined the effect of the association: CI < 0.9: synergy; 0.9 ≤ CI < 1.1: additive effect; CI > 1.1: antagonism.
Statistical analysis
Statistical analysis was performed using Wilcoxon–Mann Whitney. Statistically significant differences were defined by a p value lower than 0.05.
Results
Inhibition of PGE2 production in RAW 264.7 murine macrophages
Inhibition of COX-2-catalyzed PGE2 production from LPS-induced RAW 264.7 cells by 2’-hydroxychalcones.

aAll compounds were used at 10 μM.
b% inhibition = 100×[1-(PGE2 of LPS with the 2’-hydroxychalcones treated group)/PGE2 of LPS treated group].
cNS-398, N-(2-cyclohexyloxy)-4-nitrophenylmethanesulfonamide, was used as the reference compound. All values represented here were arithmetic mean of triplicate.
dNot determined. A promoting effect of PGE2 production was observed.
In this study, 2’-hydroxychalcone (1), whose PGE2 inhibitory activity was previously described, exhibited 80.2% inhibition on PGE2 production. Surprisingly, 2’-hydroxy-4-methylsulfonylchalcone (2) was found to induce PGE2 production. Compound 3 bearing the methylsulfonyl pharmacophore on the B ring in association with a 4’-methyl group on the A ring presented a significant inhibitory effect against PGE2 production. And, 4’-chloro-2’-hydroxy-4-methylsulfonylchalcone (4) displayed a lower activity against PGE2 production compared with 2’-hydroxychalcone 1. In the same way, 2’-hydroxy-4-sulfamoylchalcone (5), with the COX-2 sulfonamide pharmacophore on the B ring, presented a lower inhibitory effect than 2’-hydroxychalcone 1, whereas compounds 6 and 7 possessing this sulfonamide moiety on the B ring in conjunction with, respectively, a 4’-methyl and a 4’-chloro on the A ring were found to be potent inhibitors of PGE2 production. Therefore, these results demonstrated that the ability of the para-sulfonamide or methylsulfone to be a COX-2 pharmacophore was sensitive to the nature of the other phenyl substituents.
Finally, 4’-chloro-2’-hydroxy-4-sulfamoylchalcone (7) showed the best potency against PGE2 production among the whole series of 2’-hydroxychalcones with 88.9% inhibition; this result comes closer to the inhibition percentage (100%) of NS-398.
Besides, all tested compounds showed no or less than 10% reduction of MTT assay, indicating that they were not significantly cytotoxic to RAW 264.7 murine macrophages by presence or absence of LPS. Therefore, the inhibition of PGE2 production from these cells by the 2’-hydroxychalcones could not be associated with a cytotoxic mechanism.
Compounds 1, 3, 6, and 7 were then selected for the following assays since they demonstrated a significant activity against PGE2 production.
Cytotoxicity assay
Cytotoxic concentration 50% (CC50) and 90% (CC90) determined for chalcones 1, 3, 6, 7, indomethacin, and ganciclovir in growing and confluent MRC-5.
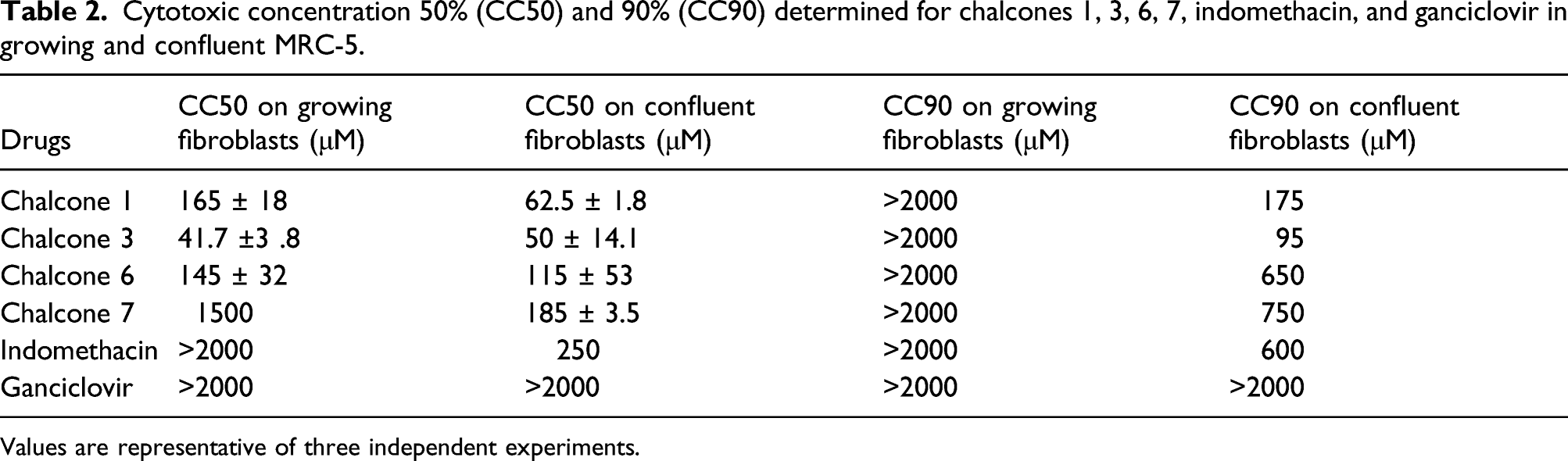
Values are representative of three independent experiments.
Impact of molecules on PGE2 production in MRC-5 fibroblasts
As shown in Figure 2, we observed a 4.3-fold increase of PGE2 amount in infected cells compared to control. Adding chalcones 7, 6, and 1 to infected cells slightly decreases the amount of PGE2 (1.2- to 1.5-fold). We observed no impact for chalcone 3 and GCV. A strong decrease was observed with indomethacin added to infected cells (17.6-fold). Chalcone 1 and indomethacin decreased the amount of PGE2 in non-infected cells (2.2-fold and 1.5-fold, respectively). Amount of PGE2 on MRC-5 cells supernatants infected with HCMV or not depending from the different chalcone derivatives used. Mock: control without compound. AD169: Infected cells with the laboratory strain HCMV AD169. GCV: Ganciclovir. Error bars represent SD values from n = 3 independent experiments.
Antiviral activity of chalcones derivatives
Effect of COX-2 inhibitors on clinical strains CHAJ06 (resistant strain with UL97 C592G+UL54 N408K G481A mutations), JAMP01 (resistant strain with UL97 A595S), and TROA07 (congenital naïve).

Mean ± SD values are shown for the active compounds from n = 3 independent experiments. EC50= efficacy concentration 50%; SI= Sensitivity Index [EC50 (clinical strain)/EC50 (AD169)]; TI50=Therapeutic index (CC50 on confluent fibroblasts/EC50).
The EC50 of chalcones and indomethacin was also determined for clinical HCMV strains: two multidrug resistant isolates (JAMP01 and CHA J06) and one naïve congenital isolate (TROA07), in comparison with GCV. We confirmed the resistance of JAMP01 and CHAJ06 strains to GCV (SI =12.4 and 4.4, respectively). All clinical isolates were susceptible to the chalcones and to indomethacin (SI < 3 for all isolates).
Inhibition of the early stages of viral cycle
We analyzed expression of HCMV immediate early proteins (IE1-72 and IE2-86) in presence of the new COX-2 inhibitors and indomethacin by western blotting (Figure 3). Due to its toxicity, we observed a lack of proteins with chalcone 3. IE1-72 and IE2-86 were expressed in the control (infected cells without compound). GCV, used as a control for antiviral activity, and all compounds except chalcone 3 decreased IE1-72 production. A decrease of both IE1-72 and IE2-86 proteins was observed only for chalcone 1 and indomethacin. HCMV immediate-early protein production in presence or absence of the different COX inhibitors. +: presence of compound/AD169; -: absence of compound/AD169; GCV: ganciclovir; Indo: indomethacin; B-actin: beta actin as a control.
Association of chalcone 7 with other antiviral compounds
Molecules in association in vitro.

Dm (dose–response values median effect (EC50)), m (shape of the dose–response curve), and r (linear correlation coefficient; r=1 indicates perfect fit) for each single drug were derived using Compusyn software (Chou and Martin, 2007). Based on these values, the CI was derived for each drug combination. CI < 0.9, synergy; 0.9 ≤ CI < 1.1, additive effect; CI > 1.1, antagonism. Values are representative of three independent experiments. Indo: indomethacin; GCV, ganciclovir; MBV, maribavir; BAI, baicalein; QUER, quercetin; ART, artesunate.
Association of chalcone 7 was synergistic with MBV or BAI, additive with GCV or ART, and antagonistic with QUER. Concerning the non-specific cyclooxygenase inhibitor indomethacin, only the association with MBV was synergistic; adding flavonoids or ART resulted to antagonistic effect, and finally, association with GCV was additive.
Discussion
To enhance its replication, HCMV increases amount of COX-2 enzyme in infected cells and establishes an inflammatory state. 10 Moreover, the association of a specific anti-COX-2 drug with a CMV inhibitor is known to enhance the inhibitory effect on viral replication. 27 This may represent an opportunity for adjuvant therapy of difficult-to-treat diseases associated with HCMV, severe brain tumors such as medulloblastoma in children, 33 or eventually glioblastoma. However, COX-2 inhibitors have various side effects, and there is a need for safer drugs. In the present study, seven chalcones were tested for their activity against COX-2-catalyzed PGE2 production; four of them were selected to be tested against HCMV infection in vitro because they demonstrated a significant activity against PGE2 production (chalcone derivative compounds 1, 2, 6, and 7).
Regarding the toxicity of these compounds, all of them were more toxic than ganciclovir and indomethacin, but with various levels. Compounds 6 and 3 have substituents found in coxibs like celecoxib (CH3 and SO2NH2) and etoricoxib (SO2CH3 and CH3). 26 The SO2NH2 group seems to increase the toxicity of chalcone 3 compared to 1. Chalcone 7 was the less toxic compound; the presence of a chlore atom seems to reduce toxicity compared to compound 6, which has a CH3 group. However, they are much less toxic than other chalcones (tri-hydroxychalcones) we have previously tested. 24
We demonstrated that these molecules have an antiviral efficacy similar to the non-specific COX inhibitor indomethacin. Moreover, all those compounds are effective at the same level against clinical strains, multi-drug resistant, and naïve congenital strains.
As expected, all the compounds act before the early stage of the viral cycle, demonstrated by the decreased production of IE proteins. A concentration of 2×EC50 was used to limit potential bias of toxicity. Use of chalcone derivative compounds impacts IE1 synthesis strongly and in variable way IE2 synthesis depending on the drug. Anti-viral efficacy of chalcone 3 cannot be interpreted due to its high toxicity, proved by the low level of actin detected on the Western Blot. As for their impact on PGE2 production in infected cells, the chalcones have a weak impact compared to indomethacin. As a whole, our data suggest multiple targets for these drugs in agreement with the variable antiviral efficiency of the different chalcone derivatives depending also on the viral strain.
The very early inhibition is very interesting for combination with other anti-viral compounds targeting DNA replication or late stages of the viral cycle. Association of a specific COX-2 inhibitor and an anti-CMV compound is known to significantly impair CMV replication. 27 Therefore, we chose to combine chalcone 7, which was the less toxic and the most efficient new anti COX-2 compound, with other antiviral molecules. Interestingly, chalcone 7 is more often synergistic or additive than indomethacin. The difference in the mode of action and the structure could explain this behavior. Association of chalcone 7 with quercetin, which inhibits early HCMV proteins synthesis, 24 is antagonistic, probably due to their similar chemical structure, and to the inhibition of COX-2 by quercetin previously described. 34 Surprisingly, the association between chalcone 7 and artesunate is additive, while we could expect a synergistic combination as artesunate targets the NF-kB pathway. 35 However, the anti-malaria molecule has a short half-life, which could explain the additive relationship and the impossibility to reach a synergistic effect. 36 The other associations are synergistic, which is hopeful for an eventual use of specific COX-2 inhibitors with more conventional anti-HCMV treatments in combination therapy, to avoid resistant mutations emerging and to reduce concentrations of administered drugs in case of side effects. The synergy with maribavir was expected, as this new drug acts at the late stage of the viral cycle by inhibition of the nuclear egress mediated by the viral kinase UL97. 37 We previously demonstrated synergy of MBV with ART and additive effect with BAI, an immediate early stages inhibitor. 38 This could be of interest to prevent the emergence of resistance. Even letermovir, the last antiviral developed targeting the terminase complex, has already shown some limits with a weak genetic barrier leading to rapid emergence of resistance mutations39,40 and could be further tested. In this context, chalcones, targeting cellular proteins, should stay efficient (as it is the case for artesunate), on resistant viruses and could act as an adjuvant for direct anti HCMV drugs.
Although development of COX-2 inhibitors with a chalcone core is a promising proof of concept, and confirms anti-COX-2 antiviral potential, additional studies are necessary in order to better characterize their bioavailability and their toxicity. Further studies may be necessary to identify new compounds with less toxicity and the same efficacy.
Supplemental Material
sj-pdf-1-avt-10.1177_13596535211064078 – Supplemental Material for Impact of new cyclooxygenase 2 inhibitors on human cytomegalovirus replication in vitro
Supplemental Material, sj-pdf-1-avt-10.1177_13596535211064078 for Impact of new cyclooxygenase 2 inhibitors on human cytomegalovirus replication in vitro by D Andouard, R Gueye, S Hantz, C Fagnère, B Liagre, L Bernardaud, C Pouget, JL Duroux and S Alain in Antiviral Therapy
Supplemental Material
sj-pdf-2-avt-10.1177_13596535211064078 – Supplemental Material for Impact of new cyclooxygenase 2 inhibitors on human cytomegalovirus replication in vitro
Supplemental Material, sj-pdf-2-avt-10.1177_13596535211064078 for Impact of new cyclooxygenase 2 inhibitors on human cytomegalovirus replication in vitro by D Andouard, R Gueye, S Hantz, C Fagnère, B Liagre, L Bernardaud, C Pouget, JL Duroux and S Alain in Antiviral Therapy
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Inserm and the National Reference Center for Herpesviruses (Santé Publique France).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.