
Abstract
Objective
The bioequivalence of the simplified protease-inhibitor-based HIV-1 antiretroviral regimen darunavir/cobicistat (DRV/COBI) 600/90-mg fixed-dose combination (FDC) tablet dispersed in water was evaluated in healthy adults and swallowability in children living with HIV aged >3 years and weighing ≥15 to <25 kg, respectively.
Methods
In the bioequivalence study 32 healthy adult participants received either a single oral dose of the DRV/COBI-600/90-mg FDC tablet dispersed in water (test) or the separate formulations (DRV 100-mg/mL at a dose of 600-mg and COBI 90-mg tablet: reference) separated by ≥ 7 days of washout. In a separate acceptability study children living with HIV-1, aged ≥3 years and weighing ≥15 to <25 kg, received a single oral dose of the dispersed DRV/COBI-600/90-mg FDC tablet. Acceptability questionnaires were completed by observers, participants and caregivers.
Results
The bioequivalence study indicated that the geometric mean ratios for DRV maximum plasma concentration and area under the concentration–time curve of the dispersed DRV/COBI-600/90-mg FDC tablet versus the separate formulations fell within the 80–125% bioequivalence limits. In the acceptability study in children, per independent observers 83% (10/12) of the children were able to swallow the dispersion completely and rated the dispersed FDC tablet as “ok” to “very easy” to swallow.
Conclusion
The DRV/COBI 600/90-mg FDC tablet dispersed in water was bioequivalent to coadministration of the separate formulations and was acceptable for long-term daily use in the intended pediatric population.
Introduction
Antiretroviral therapy (ART) has considerably decreased HIV-related morbidity and mortality.1,2 HIV infection is a manageable chronic condition that requires life-long treatment with ART. However, sustained adherence to ART is the cornerstone for achieving effective viral suppression, improved health, minimizing development of drug resistance, and reducing HIV transmission.3–6 Simplified ART regimens, such as once-daily, single-tablet, fixed-dose combination (FDC) regimens, have advantages over the traditional multi-drug regimens by providing enhanced convenience and a reduced pill burden.7–10 The pediatric population has additional challenges, that includes medication swallowability and palatability, which can negatively impact treatment adherence.11–14 Thus, there is a need to develop simplified ART in weight-adjusted and age-appropriate formulations for effective treatment of the pediatric population living with HIV.
Darunavir/cobicistat (DRV/COBI), single-tablet is a protease-inhibitor-based simplified ART regimen. The DRV/COBI 800/150-mg and DRV/COBI 675/150-mg FDCs, administered orally once daily in combination with other antiretrovirals (ARVs), have been approved for the treatment of HIV-1 in treatment-naïve and treatment-experienced patients: DRV/COBI 800/150-mg FDC for adults and pediatric patients weighing ≥40 kg (US, EU) and >12 years (EU) and DRV/COBI 675/150-mg FDC in pediatric patients weighing ≥25 to <40 kg (US). 15 The Bioequivalence of the DRV/COBI 800/150-mg FDC tablet relative to coadministration of the separate commercially available formulations has been demonstrated in healthy adults. 16
An oral DRV/COBI 600/90-mg FDC tablet for dispersion is under development for the pediatric population aged ≥3 years and weighing ≥15 to <25 kg. A dispersible tablet formulation is a practical, convenient dosing option that can help improve treatment adherence in this age group who may find it difficult to swallow bulky tablets. 13 The bioequivalence of DRV 600 mg in the presence of COBI 90 mg, when administered as a single dose of the DRV/COBI 600/90-mg FDC tablet dispersed in water compared to coadministration of the separate formulations was assessed in healthy adult participants, and the acceptability of the dispersed DRV/COBI 600/90-mg FDC tablet was evaluated by children living with HIV.
Methods
Study design
Bioequivalence study (EudraCT number: 2021-003955-40)
This was a phase 1, open-label, randomized, 2-way cross-over study conducted to evaluate the single-dose pharmacokinetics (PK) and bioequivalence of the DRV/COBI 600/90-mg FDC tablet (sourced from Johnson & Johnson) dispersed in water compared to coadministration of the separate formulations, DRV suspension (100-mg/mL at a dose of 600-mg, (sourced from Johnson & Johnson) and COBI 90-mg tablet (sourced from Gilead). This study was conducted between June 2022 and October 2022 at the SGS Belgium N.V., Clinical Pharmacology Unit, Edegem, Belgium. The study consisted of 3 phases and the total duration per participant was ∼7 weeks: a screening phase (⁓4 weeks; (days −28 to −2); an open-label treatment phase consisting of 2 single-dose treatment periods of 4 days each (days 1–4), each separated by a washout period of at least 7 days between dosing (starting on day 1); and an end-of-study/follow-up assessment phase (7–10 days after final dosing).
Before initiating any treatment, the participants were randomized as per computer-generated schedule to one of the two treatment sequences (test-reference and reference-test). Participants received both the treatments during each of the two treatment periods, as per the assigned sequence: either a single oral dose of the DRV/COBI 600/90-mg FDC tablet dispersed in water (test treatment), or single oral dose of 6 mL of DRV suspension corresponding to DRV 600-mg and a COBI 90-mg tablet (reference treatment). The study utilized a 2-way cross-over design wherein each participant served as his/her own control. During each treatment period, participants stayed at the study center for the administration of study drug(s) and blood sample collection for PK assessments. Participants fasted overnight for at least 10 h and took the study treatment on day 1 of each period, within 30 min of starting a standardized high-fat breakfast (Supplemental Methods). For each individual participant, the washout period between subsequent intakes of study drug(s) was at least 7 days, with day 1 of a treatment period considered as the first day of the washout period. Participants returned to the study center for either treatment period 2 or end-of-study/follow-up assessments, as applicable.
Acceptability study (NCT05197075)
This was a phase 1, open-label, single-dose, multicenter study to assess the acceptability of the DRV/COBI 600/90-mg FDC tablet (sourced from Johnson & Johnson) dispersed in water. The study was carried out across the Republic of South Africa (2 centers), Spain (3 centers), and the USA (1 center) with a total duration of ∼5 weeks per participant: a screening phase (∼3 weeks; days −21 to −1), an open-label drug administration phase (day 1), and a follow-up phase (8–11 days postdose). Participants were to continue their ARV regimen without interruption or change in administration schedule. On day 1, participants took a single oral dose of the DRV/COBI 600/90-mg FDC tablet dispersed in water (Supplemental Methods). Acceptability questionnaires were completed by participants, caregivers and observers within 15 min of taking the dispersion.
The Institutional Review Board and Ethical Committee approved the protocols and other study relevant documents. The studies were carried out in compliance with the International Conference on Harmonization recommendations on Good Clinical Practice and the Declaration of Helsinki. Participants, their parent(s), or their legally accepted representative provided signed informed consent, and children ≥7 years of age provided their assent for participation in the study.
Participants
The bioequivalence study included healthy adult participants aged 18–55 years with a body weight of ≥50.0 kg and body mass index (BMI) between 18.5 and 30.0 kg/m2, and considered healthy based on physical examination, medical and surgical history, clinical laboratory tests, 12-lead electrocardiogram (ECG) and vital signs. Key exclusion criteria were a history of, or current clinically significant medical or psychological illness or malignancy; clinically significant laboratory abnormalities; any active infection; a history or current use of drugs of abuse, alcohol or nicotine-containing substances.
The acceptability study included children living with HIV-1, aged ≥3 years and weighing ≥15–<25 kg, with viral load <400 copies/mL within 6 months of screening, who were on stable ARV regimen for ≥3 months prior to screening. Key exclusion criteria were active oral infection or other physical or psychological conditions that could prevent the child from swallowing or potentially confound the study assessments and outcomes.
Assessments
Pharmacokinetics and bioequivalence
For each treatment period, blood samples for PK analysis were collected for up to 72 h (time points: 0, 2, 4, 6, 8, 12, 18, 24, 36, 48 and 72 h) postdose. The primary PK parameters included maximum plasma concentration (Cmax), area under the plasma concentration–time curve from time 0 to time of the last quantifiable concentration (AUClast), and area under the plasma concentration–time curve from time 0 to infinity (AUC∞) for DRV, the active drug, in test and reference treatments. The secondary PK parameters included Cmax, AUClast, and AUC∞ for COBI, the PK booster. The plasma concentrations of both the drugs were determined using a validated, specific, and sensitive high-performance liquid chromatography-tandem mass spectroscopy with a lower limit of quantification of 5.00 ng/mL.
Safety
Safety and tolerability assessments included evaluation of treatment-emergent adverse events (TEAEs), clinical laboratory values, electrocardiograms, vital signs, and physical examination. The TEAEs were reported as per the Medical Dictionary for Regulatory Activities (MedDRA, version 20.0) coding. The severities of TEAEs were graded using the Common Terminology Criteria for Adverse Events (CTCAE, version 4.03).
Acceptability
The acceptability of the DRV/COBI 600/90-mg FDC tablet dispersed in water was assessed based on three questionnaires. The primary endpoint was a study site observer-filled questionnaire to evaluate the participants’ ability to swallow the dispersed FDC tablet either fully, partially or not-at all. The secondary endpoints were, i] a participant-filled questionnaire to evaluate the ease of swallowing, palatability and acceptability of daily intake of the dispersed FDC tablet, and ii] a caregiver-filled questionnaire to evaluate ease of swallowing, palatability, acceptability of daily intake of the dispersed FDC tablet and the ease of dispersion. For the participant and caregiver acceptability questionnaires, a 5-point hedonic scale with facial expressions to depict likeability or difficulty level was used (Supplemental Methods).
Statistical analysis
Bioequivalence study
A minimum of 32 participants were planned for this study (sample size calculation in Supplemental Methods).16,17 The PK parameters were estimated by noncompartmental analysis using Phoenix WinNonlin Version 8.1 (Certara L.P.). All statistical analyses were performed using SAS Version 9.4 (SAS Institute Inc.). All the data were summarized using descriptive statistics. The bioequivalence of test and reference treatments was evaluated using mixed-effect model to estimate the least squares means and intra-individual variance, the point estimate and 90% confidence interval (CI) of the geometric mean ratios (GMRs) (Supplemental Methods). The two treatments were considered bioequivalent if the 90% CI of the GMRs for the primary DRV PK parameters fell within the predefined bioequivalence limit of 80.0–125.0%.
Acceptability study
A target of 12 participants was to be enrolled in this study (sample size calculation in Supplemental Methods). For the assessment of the ability to swallow, the proportion of participants able to swallow the dispersed FDC tablet “fully” versus “partially” or “not at all” was reported along with a corresponding 95% CI calculated using Wilson method. For other secondary acceptability assessments, frequencies per response-category were tabulated.
For safety analysis, the incidences of TEAEs overall and by study treatments were reported for both the studies.
Analysis sets
The PK analysis set for the bioequivalence study included all participants who received ≥1 dose of study drug and had ≥1 plasma concentration data value or ≥1 PK parameter value after drug administration. For the acceptability study, intent-to-treat (ITT) analysis set was used which included all participants who were enrolled and received at least partial study intervention. For both the bioequivalence and acceptability study, the safety analysis set included all participants who received ≥1 dose of study drug.
Results
Bioequivalence study with DRV/COBI 600/90-mg FDC tablet
Participants
Of the total 32 healthy participants (16 participants per treatment sequence) enrolled in the study, 30 (93.8%) completed the study (n = 1 participant tested positive for COVID-19 in between the two treatment periods and discontinued the study; n = 1 participant withdrew consent). Overall, the mean (SD) age of participants was 39.2 (9.4) years, majority were female (56.3%), White (96.9%) and the median (range) BMI was 23.4 kg/m2 (18.9 to 29.2 kg/m2; Table S1).
Pharmacokinetic parameters
All 32 participants were included in the PK analysis set. The mean plasma concentration–time profiles of COBI and DRV were similar following administration of the test and reference treatments (Figure 1). Mean (SD) plasma concentration–time profiles for (A) DRV, and (B) COBI after oral administration of DRV/COBI 600/90-mg FDC tablet or coadministration of the separate formulations (DRV 100-mg/mL suspension at a dose of 600-mg and COBI 90-mg tablet). COBI: cobicistat; DRV: darunavir; FDC: fixed-dose combination; PK: pharmacokinetics; SD: standard deviation.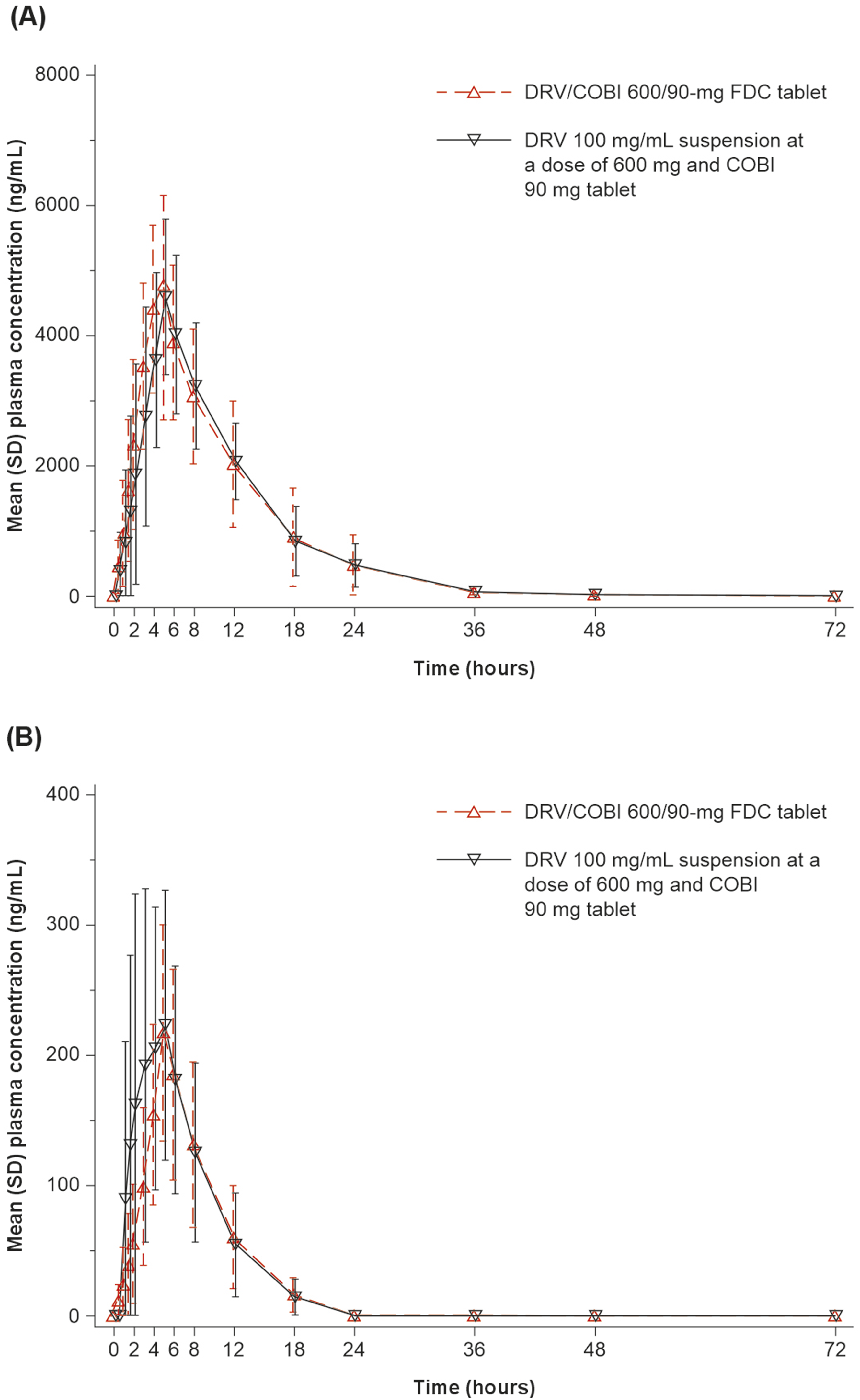
Pharmacokinetic parameters for DRV and COBI after oral administration of DRV/COBI 600/90-mg FDC tablet or coadministration of the separate formulations (DRV 100-mg/mL suspension at a dose of 600-mg and COBI 90-mg tablet).

Data are presented as mean (SD) for Cmax, AUClast, AUC∞, t1/2 and as median (range) for tmax.
AUC∞: area under the plasma concentration–time curve from time zero to infinity; AUClast: area under the plasma concentration–time curve from time zero to last measurable concentration; Cmax: maximum plasma concentration; COBI: cobicistat; DRV: darunavir; FDC: fixed-dose combination; SD: standard deviation; t1/2: terminal half-life; tmax: time to Cmax; tlast: time to last measurable concentration.
an = 31 for AUC∞, t1/2.
bn = 30 for AUC∞, t1/2.
Summary of the statistical analysis of the pharmacokinetic parameters for DRV and COBI after oral administration of DRV/COBI 600/90-mg FDC tablet or coadministration of the separate formulations (DRV 100-mg/mL suspension at a dose of 600-mg and COBI 90-mg tablet).

AUC∞: area under the plasma concentration–time curve from time zero to infinity; AUClast: area under the plasma concentration–time curve from time zero to last measurable concentration; Cmax: maximum plasma concentration; COBI: cobicistat; CI: confidence interval; CV: coefficient of variance; DRV: darunavir; FDC: fixed-dose combination; GMR: geometric mean ratio.
Safety
Both DRV/COBI 600/90-mg FDC tablet dispersed in water and the separate formulations (DRV 100-mg/mL suspension at a dose of 600-mg and COBI 90-mg tablet) showed no tolerability concerns in the healthy adult participants. Overall, 13 (40.6%) participants experienced at least one TEAE during the two treatment periods. No TEAEs leading to interruption or discontinuation of study participation or study intervention were reported. Most (9 [29%]) of the TEAEs were of grade 1 in severity. Grade 2 TEAEs were observed in 2 (6%) participants following administration of the FDC tablet, and 2 (7%) participants following coadministration of the separate formulations. No grade 3 or grade 4 TEAEs were reported. No serious TEAEs or TEAEs leading to death were reported during the study. Diarrhea was the most common TEAE (>15%, overall); all other TEAEs were reported for only 1 (3%) participant (Table S2).
Acceptability of DRV/COBI 600/90-mg FDC tablet dispersed in water
Participants
All 12 (100%) children enrolled in the study completed the study. The mean (SD) age was 5.6 (1.4) years; 3 (25%) children were ≥3 to <5 years of age and 9 (75.0%) were ≥5 years of age. The median (range) weight was 22.7 kg (15.1–23.9 kg); 4 (33%) children were in the ≥15 to <20 kg weight group (Table S3).
Observer-assessed acceptability
Observers recorded that 10 of 12 participants (83%) were able to swallow the FDC tablet dispersed in water fully (95% Wilson CI: 75.75–100.00). Of the 2 participants who swallowed the dispersion partially (both females), 1 participant gagged and experienced a bad taste and 1 participant refused to take the complete dispersion.
Participants-assessed acceptability
Most participants found it “ok” to “very easy” to swallow the FDC tablet dispersed in water (10/12; 83%) and rated daily intake as “not sure” to “like a lot” (8/12; 67%). The majority of participants (8/12; 66%) reported “like a little” to “like a lot” for the taste of the FDC tablet dispersed in water (Figure 2). Participant responses to questionnaires (A, B, and C) assessing the acceptability of the DRV/COBI 600/90-mg FDC tablet dispersed in water. COBI: cobicistat; DRV: darunavir; FDC: fixed-dose combination. aThis response was not recorded for 1 participant. Note: numbers have been rounded and therefore might not add up to 100%.
Caregiver-assessed acceptability
Caregiver responses were aligned with those from the participants, with the majority reporting that it was “ok” to “very easy” for their child to swallow the FDC tablet dispersed in water (10/12; 83%) and daily administration would be “ok” to “very easy” (11/12; 92%). Half of the caregivers (6/12; 50%) thought their child perceived the taste of FDC tablet dispersed in water as “not sure” to “like a lot.” Dispersion of the tablet in water was rated as “ok” to “very easy” by most caregivers (10/12; 83%) (Figure 3). Caregiver responses to the questionnaires (A, B, C, and D) assessing the acceptability of the DRV/COBI 600/90-mg FDC tablet dispersed in water: Participant response. COBI: cobicistat; DRV: darunavir; FDC: fixed-dose combination.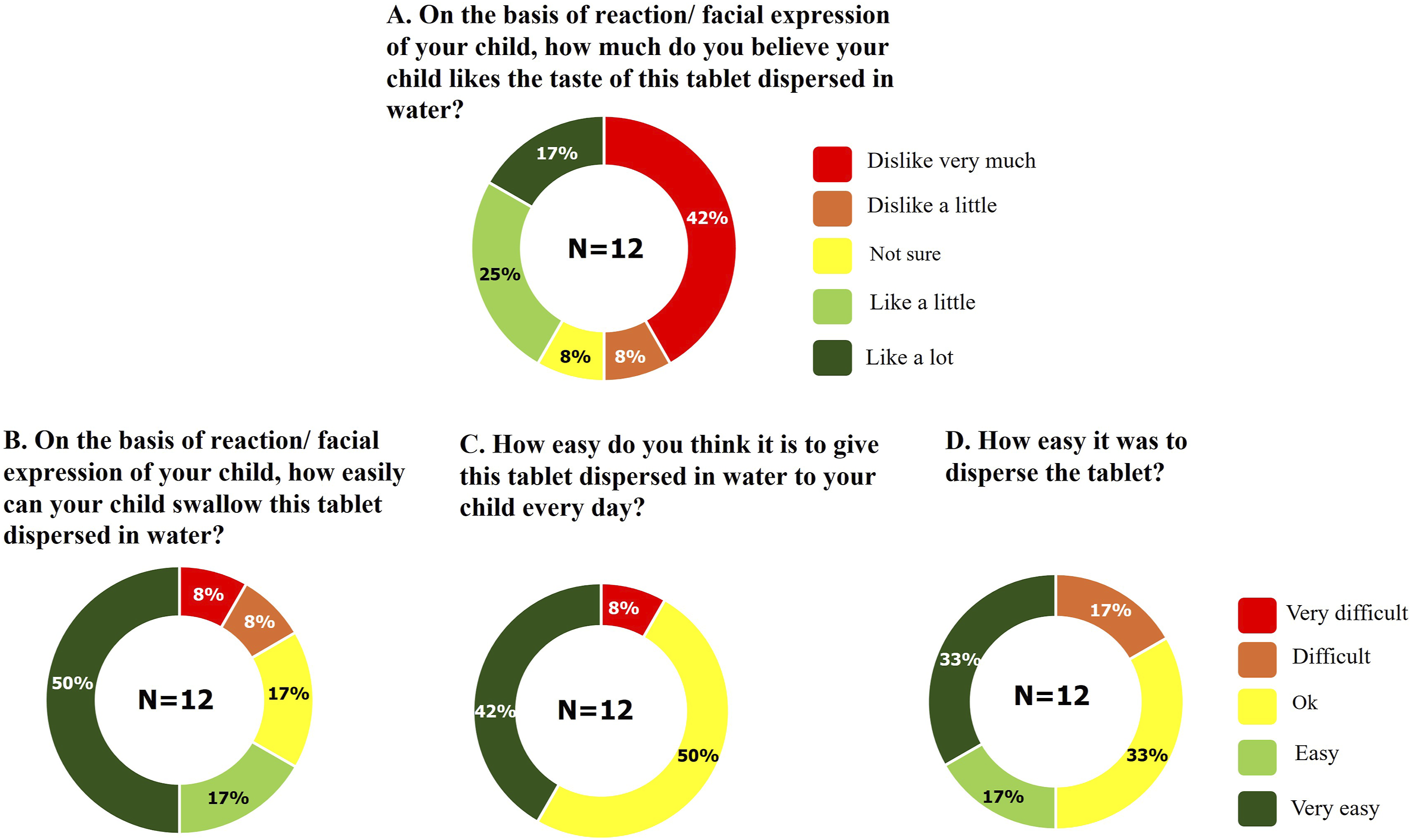
Safety
A single dose of the DRV/COBI 600/90-mg FDC tablet dispersed in water showed no tolerability concerns and no TEAEs were reported in the children who participated in this study.
Discussion
In two separate clinical studies (1) the bioequivalence of the DRV/COBI 600/90-mg FDC tablet dispersed in water compared to coadministration of the separate formulations of DRV and COBI was assessed, and (2) the acceptability of the dispersed DRV/COBI 600/90-mg FDC tablet was evaluated. The DRV/COBI 600/90-mg FDC tablet dispersed in water was found bioequivalent to the separate formulations and was acceptable by children for long-term daily use. Findings of these two studies support the use of the DRV/COBI 600/90-mg FDC tablet as part of a simplified ART for effective treatment of the pediatric population living with HIV.
In the bioequivalence study, the 90% CIs of the GMRs for Cmax, AUClast and AUC∞ of DRV administered as the DRV/COBI 600/90-mg FDC tablet dispersed in water versus coadministration of the separate formulations were all within the 80.0–125.0% bioequivalence limits. COBI showed a 26% lower Cmax and 15% lower AUC∞ and AUClast when administered as the dispersed FDC tablet compared to coadministration of the separate formulations. The lower exposure of COBI when administered as part of a dispersed FDC tablet, did not result in a different PK profile of the active drug DRV, which indicates a similar boosting effect of COBI across the observed COBI levels.
Previous PK studies have established the effect of food intake on the PK of DRV and COBI administered as an FDC, with higher DRV plasma concentrations observed under fed versus fasting conditions and no significant food effect for COBI.16–18 Product labels for separate commercially available DRV and COBI formulations recommend dosing with food.19–22 Therefore, the bioequivalence of the dispersed DRV/COBI 600/90-mg FDC tablet compared to coadministration of the separate formulations was assessed under fed conditions.
The DRV/COBI 600/90-mg FDC tablet for dispersion is intended for the pediatric population living with HIV, aged >3 years and weighing ≥15 to <25 kg. Acceptability, including swallowability and palatability, of oral formulations is crucial to ensure long-term daily use in this population. In the acceptability study, as assessed by the observer, the majority of participants were able to take the dispersion fully. Also, the majority of participants and caregivers reported that the FDC tablet dispersed in water was “ok” to take” to “very easy” to swallow and rated daily intake as “not sure” to “like a lot” or “ok” to “very easy.” In addition, the taste of the FDC tablet dispersed in water and ease of dispersion were well perceived by most participants and/or caregivers. These findings are in line with studies reporting greater acceptability for and adherence to dispersible FDC tablet formulations among children, resulting into improved clinical outcomes.23,24 Child-friendly and age-appropriate formulations have been recommended by the WHO in order to assist the pediatric population in improving adherence to long-term treatments. 25 Children in the age group of 3–6 years particularly find it hard to swallow bulky tablets, and for this population dispersible tablets offer a suitable alternative mode of administration.
No new safety findings were identified with a single dose of DRV/COBI 600/90-mg FDC dispersible tablet in both healthy adults and children included in these two studies.
In conclusion, the DRV/COBI 600/90-mg FDC tablet administered to healthy adults under fed conditions was bioequivalent to coadministration of the separate formulations. Children aged ≥3 years weighing ≥15 to <25 kg living with HIV and their caregivers found that the FDC tablets dispersed in water were acceptable for long-term daily use. These findings support the use of the DRV/COBI 600/90-mg FDC tablet in the pediatric population aged ≥3 years and weighing ≥15 to <25 kg, for whom an age-appropriate simplified ART regimen is an unmet need.
Supplemental Material
Supplemental Material - Pediatric darunavir/cobicistat fixed-dose combination tablet for dispersion: Bioequivalence versus separate agents in healthy participants and acceptability in children living with human immunodeficiency virus-1
Supplemental Material for Pediatric darunavir/cobicistat fixed-dose combination tablet for dispersion: Bioequivalence versus separate agents in healthy participants and acceptability in children living with human immunodeficiency virus-1 by Sandy Van Hemelryck, Erika Van Landuyt, Sofie Deleu, Lorant Leopold, Jay Ariyawansa, Martyn Palmer and Maria Labourdette in Antiviral Therapy
Footnotes
Acknowledgments
The authors would like to express their gratitude to the participants and their families, without whom this study would not have been accomplished, and the investigational site staff for their contribution to this study. The authors also acknowledge Vera Hillewaert (Janssen Pharmaceutica NV, Beerse. Belgium) for support with bioanalysis, Uma Kundu, M Pharm CMPPTM and Jyotsana Dixit PhD (SIRO Medical Writing, Pvt. Ltd., India) for medical writing assistance, and Robert Achenbach (Johnson & Johnson, USA) for additional editorial support.
Ethical statement
The studies were approved by the relevant Ethics committees and all participants provided informed consent before participating in the studies.
Author contributions
Conception and design: All authors. Collection of data: Sofie Deleu. Data analysis and interpretation: Sandy Van Hemelryck, Erika Van Landuyt, Sofie Deleu, Lorant Leopold, Jay Ariyawansa. Manuscript writing and editing: All authors. Final approval of manuscript: All authors. Accountable for all aspects of the work: All authors.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by Johnson & Johnson.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of Johnson & Johnson and own Johnson & Johnson stock or stock options.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.