
Abstract
Background
Chronic hepatitis B (CHB) is responsible for major disease burden worldwide. However, the number of available therapies is limited; cure remains an elusive goal. JNJ-64794964 (JNJ-4964) is an oral toll-like receptor-7 (TLR7) agonist being evaluated for the treatment of CHB. Here, we investigated the capacity of JNJ-4964 to induce transcriptomic and immune cell changes in peripheral blood in healthy volunteers.
Methods
Peripheral blood was collected in the JNJ-4964 first-in-human phase 1 trial at multiple time points to assess transcriptomics and changes in frequency and phenotype of peripheral-blood mononuclear cells. Correlation of changes to JNJ-4964 exposure (Cmax) and changes in cytokine levels (C-X-C motif chemokine ligand 10 [CXCL10] and interferon alpha [IFN-α]) were evaluated.
Results
Fifty-nine genes, mainly interferon-stimulated genes, were up-regulated between 6 hours and 5 days after JNJ-4964 administration. JNJ-4964 increased frequencies of CD69, CD134, CD137, and/or CD253-expressing natural killer (NK) cells, indicative of NK cell activation. These changes correlated with Cmax, increase of CXCL10, and induction of IFN-α and were observed at IFN-α levels that are associated with no/acceptable flu-like adverse events. JNJ-4964 administration resulted in increased frequencies of CD86-expressing B cells, indicative of B-cell activation. These changes were predominantly observed at high IFN-α levels, which are associated with flu-like adverse events.
Conclusions
JNJ-4964 administration led to changes in transcriptional profiles and immune cell activation phenotype, particularly for NK cells and B cells. Together, these changes could represent a set of biomarkers for the characterization of the immune response in CHB patients receiving TLR7 agonists.
Introduction
Hepatitis B virus (HBV) is the most common chronic viral infection worldwide, with an estimated 296 million persons chronically infected and 820,000 deaths worldwide in 2019.1–3 An estimated 15% to 40% of patients with chronic hepatitis B (CHB) progress to advanced liver disease such as cirrhosis, hepatocellular carcinoma, or hepatic decompensation.4–6 Achieving functional cure, defined as hepatitis B surface antigen (HBsAg) seroclearance sustained for ≥6 months off-treatment with or without seroconversion to anti-HBs, is the current goal for emerging CHB therapeutics. 5
Barriers to functional cure include the high load of HBV antigens (e.g., HBsAg) coming from persistent HBV covalently closed circular DNA or integrated HBV genomes, impaired immune responses associated with exhausted T-cell reactivity, and the immune-tolerant environment of the liver.7–9 Stimulating pattern recognition receptors such as toll-like receptors (TLRs) can induce innate and adaptive immune responses, and TLRs represent a potential therapeutic target.10–12 TLR7 is expressed on, and agonists directly stimulate, plasmacytoid dendritic cells (pDCs), B cells, and various cells in the liver (Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells).13,14 Additionally, TLR7 agonism can indirectly lead to or further enhance stimulation of myeloid cells, natural killer (NK) cells, and T cells.15–17
JNJ-4964 (JNJ-64794964, TQ-A3334, AL-034) is an oral TLR7 agonist. In vitro, JNJ-4964 induced antiviral interferons (IFNs), pro- and anti-inflammatory cytokines, and chemokines in human whole-blood cells and peripheral-blood mononuclear cells (PBMCs).18,19 In mice infected with a recombinant adeno-associated virus carrying a replicable HBV genome, HBsAg seroconversion was observed after JNJ-4964 treatment.19,20 In a placebo-controlled phase 1 trial, JNJ-4964 as a single or multiple dose exhibited an acceptable safety and tolerability profile in healthy adults, with no serious adverse events (AEs) observed. JNJ-4964 induced interferon-stimulated genes (ISGs; including ISG15, MX1, and OAS1) in whole blood, upon minimal TLR7 engagement. In addition, JNJ-4964 elicited transient and dose-dependent induction of cytokines with potential anti-HBV activity, including C-X-C motif chemokine ligand 10 (CXCL10)/IFN-inducible protein 10 (IP-10), interleukin 1 receptor antagonist (IL-1RA), CCL2/monocyte chemoattractant protein-1 (MCP-1), and IFN-α in serum. CXCL10 was the first cytokine up-regulated at the protein level upon TLR7 engagement, and IFN-α levels <100 pg/mL were associated with no or acceptable flu-like symptoms upon TLR7 target engagement. 21
In the current study, further analysis of blood samples derived from the JNJ-4964 phase 1 study 21 was conducted. Changes over time of the whole-blood transcriptome were assessed, as were changes in frequency and phenotype of peripheral immune cell populations after JNJ-4964 treatment and their relation to JNJ-4964 exposure (Cmax) and increases in CXCL10 and IFN-α levels.
Methods
Study design and population
This was a randomized, double-blind, placebo-controlled, first-in-human phase 1 trial of JNJ-4964 in healthy adults (NCT03285620). Study design, population, pharmacokinetics (PK), and pharmacodynamics (PD) of this trial have recently been described. 21 The analysis reported here is derived from the single-ascending-dose phase of this study, in which participants received JNJ-4964 as a single oral dose of 0.2 mg (n = 6), 0.6 mg (n = 6), 1.25 mg (n = 8), or 1.8 mg (n = 6) or placebo (n = 2 per dose cohort, n = 8 total) under fasted conditions. In the food effect study, after a washout period of ≥6 weeks, participants who received 1.25 mg of JNJ-4964 or placebo under fasted conditions were given the same study drug under fed conditions.
Dosing was completed by 32 healthy adults, 1 of whom withdrew consent before entering the food effect study. Participants were mostly male (88%), White (62%), and a minority Asian (22%), as previously described. 21 Median age (29.5 years) and median body mass index (<25 kg/m2) were similar across cohorts (Supplementary Table S1).
Study assessments
Peripheral blood was collected for the evaluation of JNJ-4964 PK (Cmax), PD (transcriptomics and cytokine assessments), and PBMCs. Briefly, blood sampling for PK was performed at baseline (pre-dose), on Day 1 (0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 hours post-dosing), and on Days 2, 3, 4, 5, and 6. Blood sampling for cytokine assessments was performed pre-dose, on Day 1 (1.5, 6, and 12 hours post-dosing), and on Days 2, 3, 5, and 14. For transcriptomics, sampling was performed pre-dose and at 3, 6, and 12 hours; Days 2, 3, and 5; and week 2 post-dosing. For PBMC assessments, sampling was performed at baseline (pre-dose) and 24 hours after dosing (Supplementary Table S2).
Transcriptomic analysis
Whole blood was collected in RNA Paxgene tubes (PreAnalytiX, Hombrechtikon, Switzerland, ref. 762165). RNA was isolated using Paxgene Blood RNA kit (Qiagen, Hilden, Germany, ref. 762331) and hybridized to Clarion Go Screen gene expression microarray (ThermoFisher, Waltham, MA, USA; ref. 952361).
Data were processed using R Statistical Software (version 3.6.1, R Foundation for Statistical Computing, Vienna, Austria, https://www.r-project.org) and Bioconductor tools. 22 Gene expression values were normalized using a robust multi-array average approach. 23 The individual probes were grouped into gene-specific probe sets using ThermoFisher CDF, allowing the measurement of 19911 transcripts per sample (Supplementary Methods).
PK and biomarker analyses
PK and cytokine assessments were conducted as previously described. 21 Briefly, plasma JNJ-4964 concentrations were evaluated by a validated liquid chromatography with tandem mass spectrometry method; the lower limit of quantification (LLOQ) was 0.75 pg/mL. PK parameters were estimated using non-compartmental analysis (WinNonlin®, Certara, Princeton, NJ, USA). Serum IFN-α was assessed by an enzyme-linked immunosorbent assay (LLOQ = 12.5 pg/mL); serum CXCL10 was analysed as part of a Luminex assay panel (Austin, TX, USA; LLOQ = 25 pg/mL), and ISG15 expression in whole blood was analysed by quantitative reverse transcription-polymerase chain reaction (qRT-PCR). 21
For each participant, the Cmax, maximum fold increase CXCL10 and maximum IFN-α level was determined. Cmax is defined as the maximum concentration of JNJ-4964 (in pg/mL) measured in peripheral blood within the first week after dosing. The maximum fold increase CXCL10 was defined as the maximum fold change from baseline measured in peripheral blood within the first 96 hours post-dosing. The maximum IFN-α level was defined as the maximum concentration IFN-α (in pg/mL) in peripheral blood within the first 96 hours post-dosing.
PBMC isolation
PBMCs were isolated by centrifugation on Ficoll-Paque (GE Healthcare, Chicago, IL, USA), cryopreserved, and stored in liquid nitrogen until flow cytometric analysis was performed.
Immunostaining of PBMCs and flow cytometry data acquisition procedure
Cryopreserved PBMCs were thawed and resuspended as described in the Supplementary Methods
Statistical analysis
For transcriptomics assessments, the unsupervised analysis was based on a spectral map analysis. 24 The supervised analysis used the limma package 25 in Bioconductor/R at each time point, considering patients as a random factor. Differential analysis was performed to identify associations with gene expression and Cmax using a mixed model (Supplementary Methods). Pathway enrichment analysis was performed using Gene Ontology annotation26,27 and MLP package. 28
For each considered cell population in the PBMC flow cytometry analysis, the effect of treatment with JNJ-4964 on the magnitude of the selected cell population (percent of cells relative to parent population in the flow cytometry gating) was summarized using the value at 24 hours minus the pre-dose value (change from baseline at 24 hours). For each cell population, the analysis set contained 1 value per participant, except for participants who received 1.25 mg in the fasted/fed cohort. For those participants, 2 values were included: 1 after fasted dosing and 1 after fed dosing. Values corresponding to samples with low viability at either the pre-dose or 24-hour time point (<60% assessed by Guava Easycyte) were removed from the analysis set. For each participant, the exposure to JNJ-4964 was summarized using Cmax. In the statistical analysis, PK values below LLOQ were imputed by the LLOQ, and for participants receiving placebo, the Cmax was imputed by LLOQ. CXCL10 was summarized using the maximum fold change from baseline by 96 hours, and IFN-α was summarized using the maximum value by 96 hours, in which values below the LLOQ were imputed by the LLOQ −1. The associations between JNJ-4964 exposure (Cmax) and CXCL10 or IFN-α (all on log10 scale), on the one hand, and the change from baseline at 24 hours in the relative magnitude of the cell population, on the other hand, were quantified using the Pearson correlation coefficient. Additionally, for each cell population, whether the correlation coefficient differs from zero was tested. The reported p-values corresponding to these tests were adjusted for multiple testing using the Benjamini–Hochberg method. 29
Results
JNJ-4964 induces expression of waves of peripheral-blood genes over time, mainly comprising antiviral molecules
Substantial interindividual transcriptome variation was observed. Approximately 23% of total intersample variability in transcriptomic expression was driven by ISGs, including MX1, IFI44, IFI6, IFI44L, and dosing (1.25 mg and 1.8 mg). A dose effect on transcriptomic variability was observed, which correlated with JNJ-4964 exposure (Cmax; Figure 1). Most responses were detected 12–24 hours after dosing, with 2025/19911 transcripts interrogated significantly induced 24 hours post-dosing. The most enriched Gene Ontology biological processes observed were type I IFN response and negative regulation of viral process (Figure 1B; including up-regulation of OAS1, OAS2, IFIT2, IRF7, IRF9, RSAD2, STAT1, and USP18 and downregulation of ribosomal genes including RPL4, RPL18, RPLP0, and RPS3). Transcriptomics (absolute values) and correlation with Cmax. (
Most of the differentially expressed genes showed a comparable pattern over time, and 59 transcripts were up-regulated between 6 hours and 5 days after JNJ-4964 dosing. These included OAS1, ISG15, and MX1, already observed by qRT-PCR21 and multiple other type I IFN–related ISGs (e.g., IFIT5 and IFI44). Increased expression of IFI27 30 was gradually observed with a peak at Day 5 post-dosing (Figure 1C), indicating secondary activation of the signalling cascade in pDCs. Activation and regulation of pDCs and myeloid cell lineages were suggested by the increased expression of RSAD2 (promoting TLR7-dependent IFNβ production in pDCs); TRIM38, TRIM22, and PML (involved in macrophage activation and in the case of TRIM22, suppressed by HBV X protein (HBx) 31 ]); STAT1 and USP18 (involved in pathogen control and the negative regulation of nuclear factor κB [NF-κB] and IFN-α signaling 32 ; Figure 1D).
JNJ-4964 induces changes in peripheral immune cells
Evidence for the activation of immune cells 24 hours after single JNJ-4964 oral dosing was examined by flow cytometry. The greatest cell changes from baseline were observed for NK cells; these cells do not express TLR7, rather they are probably activated indirectly via cytokine release from other TLR7-activated immune cells. 33 The absolute percentage of CD69+ cells in the CD56+ NK cell population increased from baseline in the 1.8-mg group (percentage point increase of 61.15% ± 13.08%) as compared with placebo-treated controls (percentage point increase of 0.03% ± 5.33%; Supplementary Table S4 [rank #4]). Other activation markers also increased from baseline in the CD56+ NK cell population upon JNJ-4964 dosing as compared to placebo, including CD137 (4-1BB), CD357 (GITR), CD134 (OX40), and CD366 (TIM-3); percentage point increases of 3.59% ± 0.01%, 6.15% ± 2.19%, 0.88% ± 0.48%, and 9.60% ± 31.25%, respectively, in the 1.8-mg group; Supplementary Table S4 [rank #11, #32, #8, and #69, respectively]). Similarly, changes in monocytes, conventional DCs (cDCs; CD11c+ CD14−/dim HLA-DR+ lineage−), and B-cell subset percentages and activation status were observed; these changes, and apparent correlations with JNJ-4964 Cmax, are summarized in Supplementary Table S4. It should be noted that some of the changes observed did not always correlate well with JNJ-4964 Cmax, highlighting the complexity of responses to JNJ-4964. For example, CD366+ of NK cells (percentage point increases of 6.91%, 7.90%, and 9.60% in the 1.25-mg fasted, 1.25-mg fed, and 1.8-mg fasted groups, respectively; Supplementary Table S4 [rank #69]) and GITRL+ of CD14+ monocytes (percentage point increase of 7.00% in the 1.8-mg group only; Supplementary Table S4 [rank #49]).
Changes in frequency and activation of PBMC immune cell populations were subsequently ranked by the average p-value rank of the Pearson correlation coefficients of JNJ-4964 exposure (Cmax), post-dose CXCL10, and IFN-α protein levels (Figure 2 [top 50] and Supplementary Table 5 [all]). Interestingly, the correlations of changes in immune cell populations with CXCL10 and/or IFN-α levels were generally stronger/more significant than with JNJ-4964 Cmax (Figure 2 and Supplementary Table S5). Top 50 immune cell populations sorted by average (adjusted) p-value rank for Cmax, IFN-α, and CXCL10. Dark blue values denote p-values <0.01, light blue bars denote p-values <0.05. Dotted lines represent a Pearson correlation coefficient of ± 0.7. Cmax, maximum serum concentration; CXCL10, C-X-C motif chemokine ligand 10; IFN-α, interferon alpha. For each immune cell population, a Pearson correlation coefficient was determined separately for JNJ-4964 exposure (Cmax), post-dose CXCL10-fold change, and IFN-α protein levels. The corresponding p-values were adjusted for multiple testing (Benjamini–Hochberg) and were then ranked from smallest to largest, with the smallest p-value receiving rank 1, the second smallest rank 2, etc. The average of the 3 ranks was then calculated for each cell population, and the cell populations were sorted according to the average rank (from smallest to largest).
Increased frequency of activated circulating NK cells correlated with JNJ-4964 Cmax, CXCL10, and IFN-α protein levels
In the CD3−CD56+ NK cells, treatment with JNJ-4964 increased percentage of CD69+ (Figure 3A; median 15% point increase across JNJ-4964 doses), CD134+ (Figure 3B; median 0.17% point increase across JNJ-4964 doses), CD137+ (Figure 3C; median 1.09% point increase across JNJ-4964 doses), and CD253 (TRAIL)+ NK cells (Figure 3D). Of note, CD253 expression could only be evaluated in the 1.8-mg group because of limited availability of samples. In this dose group, a trend was observed for an increase in percentage of CD253+ NK cells (Figures 3D, 3H and 3L). Activation profile of peripheral-blood NK cells in relation to Cmax, CXCL10, ISG15, and IFN-α. 
These increases correlated with JNJ-4964 Cmax (Figures 3E–3H), CXCL10-fold increase, and IFN-α levels (Figures 3I–3L). Increased frequency of CD69+ cells coincided with the increase in peripheral ISG15 messenger RNA (mRNA), even in absence of an increase in CXCL10 (Figure 3I). Increases in CD134+ and CD137+ NK cells coincided with ≥2-fold increases in CXCL10 protein levels (Figures 3J, 3K).
In the CD3−CD56dim NK cell subset (whether CD16+ or CD16−), treatment with JNJ-4964 also increased the percentage of CD357+ cells (median 0.9% point increase across JNJ-4964 doses) and CD137+ cells (median 1.19% point increase across JNJ-4964 doses). These increases did not significantly correlate with JNJ-4964 Cmax; however, they were observed as soon as ≥2-fold increases in CXCL10 protein expression were seen (Supplementary Table S5).
No changes were observed in the frequencies of CD337+ (NKp30), CD335+ (NKp46), CD159+ (NKG2A), CD159c+ (NKG2C), HLA-DR+, CD366+ (TIM-3), perforin+, or granzyme B+ NK cells (Supplementary Table S5).
Importantly, increases in the frequency of several markers associated with NK cell activation were independent of IFN-α levels. Activated NK cells were observed at IFN-α levels above and below 100 pg/mL, a threshold that is associated with flu-like AEs. 21
Increased frequency of circulating activated B cells correlated with CXCL10 and IFN-α protein levels
Treatment with JNJ-4964 increased the percentage of circulating CD86+ B cells by 24 hours (median 0.8% point increase from baseline across JNJ-4964 doses; Figures 4A, 4B). Similar increases were observed for B-cell population subsets, namely, CD19+CD27+IgD−IgM− B cells (switched memory B cells) and CD19+CD27+IgD+ B cells (non-switched memory B cells). Similar trends were also observed for the CD27− (naive) B-cell subsets, although the correlation between exposure and cytokines was generally less strong (Figure 2, Supplementary Table S5). Changes in CD86+ B cells. (
The increase in CD86+ B cells correlated significantly with CXCL10 and IFN-α (Figure 2), most apparently for IFN-α levels >100 pg/mL (Figure 4C), which may be associated with flu-like AEs.
Increased frequency of activated monocytes correlated with CXCL10 protein levels
Treatment with JNJ-4964 induced an increased frequency of GITRL+ inflammatory CD14+CD16+ monocytes (median 0.6% point increase from baseline across all JNJ-4964 doses, Figure 5A). These increases did not correlate significantly with JNJ-4964 Cmax (Figures 3, 5B, and Supplementary Table S5) but were observed as soon as ISG15 mRNA expression was up-regulated, and before detectable levels of IFN-α (Figure 5C). This suggests that monocyte activation is attainable at JNJ-4964 doses not associated with IFN-α-driven AEs. Changes in GITRL+ CD14+CD16+ monocytes. (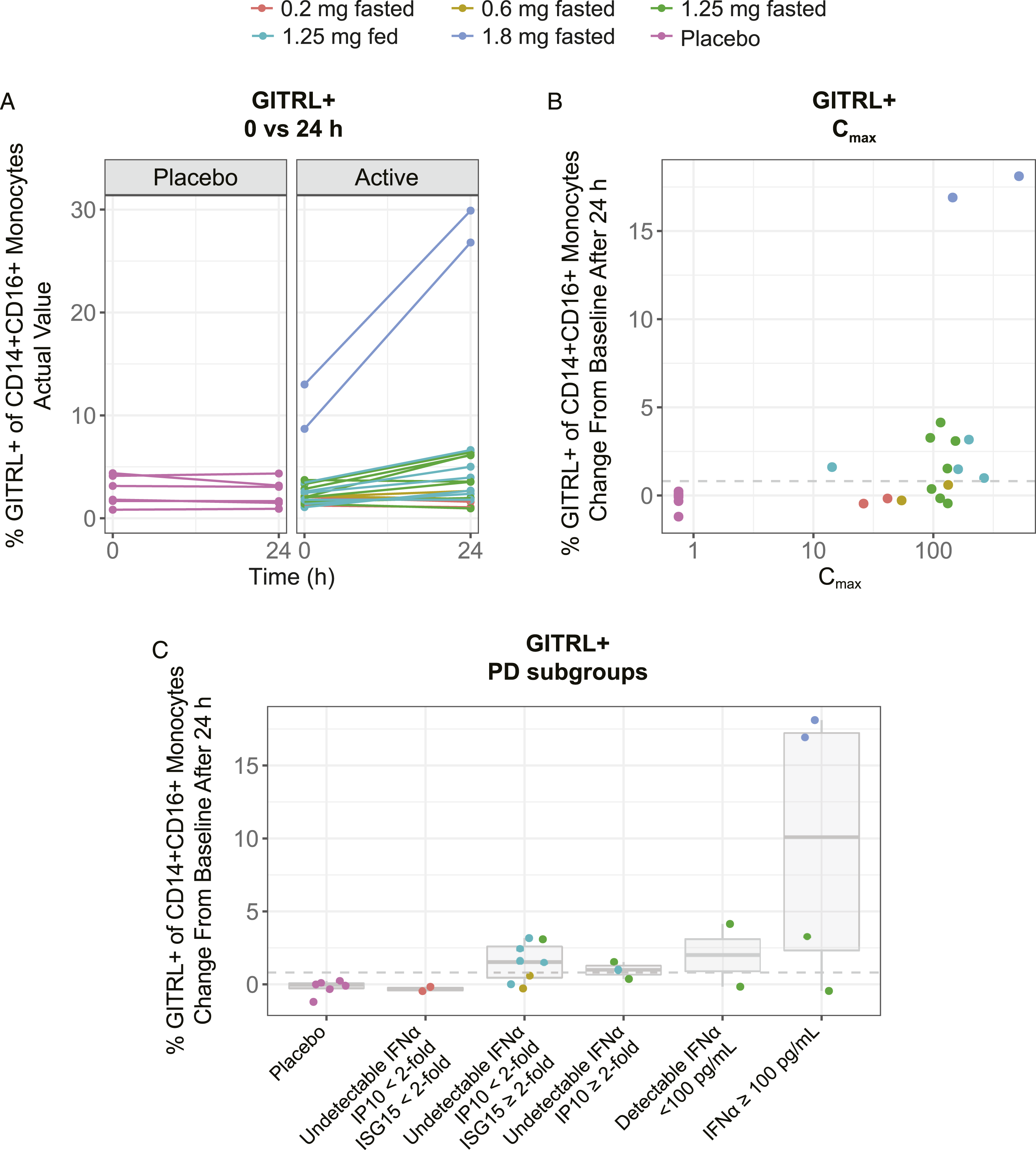
CD14+ monocytes increased in frequency from a baseline: (median 9.7% point increase from baseline across JNJ-4964 doses). This correlated with JNJ-4964 Cmax and cytokines (Figure 2 and Supplementary Table S5).
Although activation of pDCs (CD303+ CD11c− CD14−/dim HLA-DR+ lineage−) and cDCs was observed by transcriptional analysis, no changes were seen in frequencies and/or activation markers by flow cytometry (Supplementary Table S5).
Discussion
Here, we report that oral single dosing with TLR7 agonist JNJ-4964 up to 1.8 mg in healthy volunteers induced exposure-related transcriptomic and phenotypic changes on peripheral-blood immune cells that correlated with previously described induction of cytokines and ISGs. 21 We propose that immune cell activation profiles could provide a novel set of biomarkers to help characterize pharmacodynamic responses to TLR7 agonists in patients with CHB in future clinical trials.
After treatment with JNJ-4964, a TLR7-induced IFN-α signalling cascade was observed in peripheral blood, which led to the stable up-regulation of 59 transcripts from 6 hours to 5 days after dosing. These genes included antiviral ISGs such as ISG15, MX1, and OAS1, expressed by NK and T cells, among others. 30 They also included genes involved in the regulation of the NF-κB pathway (including down-regulation of ribosomal protein-coding genes, such as RPS3, which is a well-described player in NF-κB signalling). 34 Although some proteins encoded by genes identified by transcriptomics (e.g., TRIM22 35 and TRIM38 36 ) were not included in the flow cytometry panel, these are also expressed by immune cells including monocytes; our analysis of monocytes (the blood representatives of macrophages) by flow cytometry supports their activation.
Ubiquitin-specific peptidase 18 (USP18) is an ISG15-specific USP, a potent inhibitor of NF-κB and IFN-α signalling and modulator of the innate and adaptive immune system, with a variety of functions in pathogen control, cancer development, and autoimmune diseases. 32 TRIM22 is an antiviral protein; by suppressing ISGs via HBx, HBV can indirectly down-regulate the expression of TRIM22. 31 Promyelocytic leukaemia protein (PML/TRIM19) has demonstrated activity against cytomegalovirus and human immunodeficiency virus infection and is the only member of the TRIM family with in vivo evidence of antiviral activity. 37 IFI27 was significantly up-regulated at 5 days post-dosing, suggesting a secondary effect of the signalling cascade in monocyte-derived macrophages 38 and potentially pDCs (although representing a small proportion of the cells in whole blood). IFI27 is a novel immune biomarker that has been shown to have high diagnostic accuracy and specificity in discriminating between viral (influenza) and bacterial infections. 30 In chronically infected patients, HBV has been described as a stealth virus, repressing pro-inflammatory cytokines and ISG expression. 39 Several immunotherapies have been tested in humans, including TLR7 agonists,40–42 and have been shown to induce type I IFN response (measured by induction of ISG15, MX1, OAS1, as well as MHC class I and TRIM genes families, inducing activation of HBV-specific T cells and NK cells in pre-clinical models.11,43,44
Immune cell phenotyping revealed induction of molecules associated with immune cell activation in cell types that may be activated by JNJ-4964 directly (e.g., CD86 on B cells) or indirectly (e.g., CD69 and CD134 on NK cells; in particular, CD56dim NK cells). These changes correlated well with Cmax and with fold changes of ISG15, CXCL10, and IFN-α. Importantly, evidence of NK cell activation also occurred at systemic IFN-α protein levels <100 pg/mL, a threshold below which no pyrexia or lymphocytopenia was observed. 21 This suggests that a safe and tolerable profile might be attained with exposures of JNJ-4964 inducing NK cell activation in patients with CHB, with increased systemic levels of cytokines such as CXCL10 and IFN-α.
Achievement of functional cure in patients with CHB probably requires activation of innate and adaptive arms of the immune system. 45 NK cells are known to be a key innate immune cell for the control of bacterially infected, virally infected, and cancerous cells.46,47 Furthermore, circulating CD56dim NK cells are reduced in patients with CHB in the immune-active phase. 48 Thus, mechanisms by which NK cell activation/function can be increased in patients could be an important attribute of any CHB treatment regimen. NK cells probably do not express TLR7 and thus can only be indirectly activated by TLR7 agonists. 46 Here, we showed that several markers associated with NK cell activation, CD69, CD134, CD137, CD253, and CD357, were up-regulated upon dosing with JNJ-4964. One of the strongest increases from baseline was observed in CD69+ NK cells, which increased 61 percentage points. These findings suggest that NK cell activation might be used as a biomarker for oral TLR7 agonism. Increased NK cell activation was evident even in individuals who had undetectable serum IFN-α (‘pre-systemic effect’), suggesting that NK cell activation is achievable without IFN-α-associated flu-like symptoms.
Furthermore, NK cell activation was observed when ISGs and cytokines (with IFN-α levels <100 pg/mL) were both up-regulated, with the highest up-regulation observed in volunteers with IFN-α levels >100 pg/mL. The importance of CD253+ NK cells in the clearance of HBV remains somewhat controversial49,50 but may be related to timing. Following 48 weeks of pegylated IFN (PegIFN)-based treatment, HBsAg clearance was associated with increased CD253-expressing, CD56bright NK cells. 51 By contrast, a reduction in circulating HBsAg was associated with decreased CD253+ NK cells, after PegIFN and nucleos(t)ide analogue treatment. 52 Recently, early NK cell activation including CD253 expression was found to correlate with HBsAg decline in PegIFN-treated patients. 53 CD253+ NK cells have also been implicated in the depletion of vaccine-induced, HBV-specific CD8+ T cells. 54 A positive contribution of NK cells to HBV functional cure needs to be further explored and may depend upon the specific treatment regimen or CHB patient subtype.
HBsAg seroconversion is recognized as an important component for achieving HBV functional cure. Therefore, therapeutics that are also able to activate B cells could be an important consideration for any new therapeutic regimen for patients with CHB. Here, evidence of B-cell activation by CD86 up-regulation was observed within the total CD19+ B-cell population (Figures 4A, 4B). Both CD19+CD27+IgD−IgM− (switched) and CD19+CD27+IgD+ (non-switched), memory B-cell subsets showed evidence of activation. Similar trends were observed for the CD27− (naive) B-cell subsets, although these changes were generally of smaller magnitude and were not always statistically significantly associated with Cmax, CXCL10, or IFN-α (Supplementary Table 5). Whether peripheral activation of B cells is a result of direct TLR7 activation or indirect TLR7 activation via IFN-α and/or other cytokines cannot be determined by the current study. Increased frequencies of CD86+ B cells were greatest in participants with the highest serum IFN-α levels (>100 pg/mL; Figure 4C). Whereas high levels of serum IFN-α probably correlate with increased TLR7 target engagement, 55 they are probably also associated with side effects reported for TLR7 agonists such as RO7020531 (in development for CHB in combination platform trial NCT04225715) and PegIFN-α therapies, such as flu-like symptoms, haematological toxicity, and increased transaminase levels.55,56
Little to no evidence of pDC and cDC activation was observed in the current study by flow cytometry (Supplementary Table 5). However, DCs are sensitive to freeze/thaw cycles, and their abundance in these PBMC preparations was low. It is thus difficult to accurately assess their phenotype. JNJ-4964–activated pDCs and cDCs may also predominantly reside in tissue, rather than in peripheral blood. However, IFI27, a gene involved in pDC activation, was up-regulated in the transcriptomics analysis, suggesting a minimal level of activation.
How the activation of peripheral-blood immune cells upon JNJ-4964 administration relates to tissue (lymphoid/liver)-activated immune cells needs to be defined. Boeijen et al 57 reported that expression levels of ISGs and leucocyte-specific genes in liver biopsies from patients with CHB as compared to the peripheral blood were markedly different. The translatability of our findings to the liver is to be followed up in future clinical trials evaluating TLR7 agonists.
Limitations of this study include the predominantly male and White study population, the relatively small sample size of participants in each dosing cohort, and poor cell viability of some samples. The TLR7 gene is located on the X chromosome, and TLR7 agonists are associated with a stronger cytokine response in women. 58 This is consistent with previously reported variability in PD response to certain doses within participants. 58 A lower EC50 (higher responsiveness) for female sex and Asian race on ISGs and cytokines may be related to the association between TLR7 gene variants and TLR7 expression levels.58–60 The TLR7 rs3853839 C/G genetic variant is more frequent in individuals of Asian origin and may explain the higher responsiveness in Asians to TLR7 agonism.58–60 These factors need to be considered when designing future clinical trials in patients with CHB. Furthermore, results of this study were obtained in healthy individuals, and whether similar responses will be observed in CHB patients remains to be determined. Of note, other oral TLR7 agonists such as vesatolimod 61 and RO7020531 43 demonstrate comparable ISG responses between healthy volunteers and CHB patients. At the doses tested, ISG activation without cytokine (e.g., IFN-α) induction was observed in CHB patients. 62 Additionally, induction of IFN-α was not observed in healthy volunteers at the doses tested, whereas IFN-α was present at baseline in a proportion of CHB patients, indicating baseline differences may exist in CHB patients. In chimpanzees, a correlation exists between TLR7 agonist doses that induce systemic IFN-α and HBsAg decline. 63 Thus, for antiviral effect, TLR7 agonists may need to be used in combination regimens and/or doses increased to induce cytokines. Although the mode of action of JNJ-4964 is similar to other TLR7 agonists, there is potentially a larger window of opportunity for use in combination therapy. Notably, whereas ISGs and cytokine responses are typically transient, cellular activation resulting from cytokine activity is expected to be longer lasting, although still transient. In this study, we collected PBMCs at baseline and 24 hours and assessed cytokine responses up to 4 days post-dosing. 21 Following weekly administration of vesatolimod, a TLR7 agonist, evidence for NK cell activation was observed at 8, 12, and 24 weeks post-treatment. 12 The CHB patient cohorts in these studies were mostly virally suppressed, without hepatocellular carcinoma, and without coinfections and were thus likely to have limited inflammation. The effect of oral TLR7 agonism on phenotype and function of T cells, whether differential responses exist between healthy volunteers and CHB patients, and the correlation between changes in NK and B cells as biomarkers for antiviral responses remain to be formally determined for JNJ-4964.
In conclusion, JNJ-4964 induced type I IFN–associated transcriptional changes and resulted in activated phenotypes of peripheral-blood immune cells, including NK and B cells. Importantly, evidence of immune activation occurred even in the absence of systemic IFN-α elevation, suggesting that a safe and tolerable profile might be attained. The identified immune cell profiles in the current study could represent a set of biomarkers to help characterize the immune responses of patients with CHB to TLR7 agonist therapy alone and/or in combination with other anti-HBV modalities.
Supplemental Material
Supplemental Material - A single, oral dose of the TLR7 agonist JNJ-64794964 induces transcriptomic and phenotypic changes in peripheral immune cells in healthy adults
Supplemental Material for A single, oral dose of the TLR7 agonist JNJ-64794964 induces transcriptomic and phenotypic changes in peripheral immune cells in healthy adults by Wim Pierson, Marianne Tuefferd, Florence Herschke, Leen Slaets, Marjolein Crabbe, Dorien Verstappen, Steffi de Pelsmaeker, Ian Strickland, Edward J Gane, Christian Schwabe, Yingjie Zhang, Peter Meerts, Joris Vandenbossche, Pieter Van Remoortere, Inge Verbrugge and An de Creus in Antiviral Therapy
Footnotes
Acknowledgements
We express our gratitude to the participants in this study. We also thank Rod Dunbar (Auckland University) for activities related to PBMC collection for this study. This study was sponsored by Janssen Pharmaceuticals. Writing and editorial support for the development of this manuscript, under the direction of the authors, was provided by Kurt Kunz, MD, MPH (Lumanity Communications, Inc.) and funded by Janssen Pharmaceuticals.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article: WP, MT, FH, LS, MC, DV, SDP, IS, PM, JV, PVR, IV, and ADC are employees of Janssen and may be Johnson and Johnson stockholders. EJG served as an advisor and/or speaker for AbbVie, Arrowhead Pharmaceuticals, Assembly Biosciences, Gilead Sciences, GlaxoSmithKline, Janssen, Merck, Novartis, Roche, and Vir Biotechnology. CS has advised for Johnson and Johnson and Vir Biotechnology. YZ is a statistical consultant of Open Analytics, NV.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article. This work was supported by Janssen Pharmaceuticals.
Data availability statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson and Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at ![]() .
.
Trial Registration
ClinicalTrials.gov Identifier: NCT03285620.
ORCID iDs
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.