
Abstract
Background:
Paclitaxel (PTX) is touted as an essential medicine due to its extensive use as a chemotherapeutic agent for various cancers and an antiproliferative agent for endovascular applications. Emerging studies in cardio-oncology implicate various vascular complications of chemotherapeutic agents.
Methods:
We evaluated the inflammatory response induced by the systemic administration of PTX. The investigation included RNAseq analysis of primary human endothelial cells (ECs) treated with PTX to identify transcriptional changes in pro-inflammatory mediators. Additionally, we used dexamethasone (DEX), a well-known antiinflammatory compound, to assess its effectiveness in counteracting these PTX-induced changes. Further, we studied the effects of PTX on monocyte chemoattractant protein-1 (MCP-1) levels in the media of ECs. The study also extended to in vivo analysis, where a group of mice was injected with PTX and subsequently harvested at different times to assess the immediate and delayed effects of PTX on inflammatory mediators in blood and aortic ECs.
Results:
Our RNAseq analysis revealed that PTX treatment led to significant transcriptional perturbations in pro-inflammatory mediators such as MCP-1 and CD137 within primary human ECs. These changes were effectively abrogated when DEX was administered. In vitro experiments showed a marked increase in MCP-1 levels in EC media following PTX treatment, which returned to baseline upon treatment with DEX. In vivo, we observed a threefold increase in MCP-1 levels in blood and aortic ECs 12 h post-PTX administration. Similar trends were noted for CD137 and other downstream mediators like tissue factor, vascular cell adhesion molecule 1, and E-selectin in aortic ECs.
Conclusion:
Our findings illustrate that PTX exposure induces an upregulation of atherothrombotic mediators, which can be alleviated with concurrent administration of DEX. Considering these observations, further long-term investigations should focus on understanding the systemic implications associated with PTX-based therapies and explore the clinical relevance of DEX in mitigating such risks.
Keywords
Background
Paclitaxel (PTX) is a cornerstone drug in the treatment of various solid cancers such as breast, ovarian, lung, and head and neck cancers, 1 as well as in endovascular procedures for stenotic lesions. 2 It therefore has widespread use as both a systemic and a local agent, with an effectiveness in both applications that highlights the interconnectedness of cancer and cardiovascular health. Cardiovascular diseases (CVDs) are a major concern for cancer survivors, 3 often ranking as the leading cause of death following cancer recurrence or progression. Data from the Surveillance, Epidemiology, and End Results (SEER) database show that among 3,234,256 US cancer survivors between 1973 and 2012, 38% succumbed to cancer and 11.3% died from CVDs. 4 This intersection of cardiology and oncology, known as cardio-oncology, examines the cardiovascular implications of various chemotherapeutic agents, including PTX. However, there is a notable gap in the literature regarding the systemic and cardiovascular impacts of PTX, underscoring the need for further investigation in this area.
Local PTX delivery is a common component of endovascular interventions for the treatment of obstructive vascular disease, particularly peripheral artery disease (PAD). As the third leading cause of atherosclerotic morbidity, PAD affects over 8.5 million individuals aged 40 and above in the United States alone, with prevalence rising sharply among the elderly.5–7 The primary intervention for PAD, percutaneous transluminal angioplasty (PTA), 8 has evolved with the introduction of drug-coated balloon (DCB) therapy, especially using PTX as an antiproliferative agent. Historically, the use of PTX-coated devices in vascular interventions has been a topic of extensive debate, primarily due to concerns regarding its long-term safety. 9 Recent developments, however, have significantly altered the landscape of PTX use in cardiovascular medicine. The United States Food and Drug Administration (FDA) updated its stance in July 2023, based on comprehensive reviews of randomized trials and extensive data analyses. This update has downplayed the earlier warnings about the mortality risks associated with PTX-coated devices,10,11 indicating a shift in the understanding of PTX’s safety profile in endovascular treatments. Despite these regulatory changes, the potential remains for (unintended) circulatory release of PTX in PAD treatment due to intraprocedural drug wash-off from DCBs and/or treated vascular sites.
The systemic effects of PTX, particularly in the context of its broader application in oncology and PAD, warrant further exploration. Our study aims to delve into the systemic effects of PTX in cell culture and mouse models, hypothesizing that increased systemic exposure to PTX could trigger a cascade of biochemical events altering cytokine signaling and pro-inflammatory pathways. Our research also explores compensatory strategies to minimize potential PTX-induced systemic toxicity. This approach is pivotal considering the FDA’s recent reassessment, as it could contribute to reinvigorating the clinical use of PTX-coated devices while ensuring patient safety.
Methods
Here we investigated the inflammatory response triggered by systemic administration of PTX and explored potential strategies to mitigate these effects. We conducted RNA sequencing analysis on primary human endothelial cells (ECs) treated with PTX. We further validated these transcriptional changes in EC lysates. To assess the immediate and delayed effects of PTX, we analyzed a cohort of mice at various times following PTX injection.
Study design
All animal experiments were conducted after IACUC approval from Boston University and Boston University Medical Campus. All procedures conformed to the guidelines from Directive 2010/63/EU of the European Parli-ament on the protection of animals used for scientific purposes or the NIH Guide for the Care and Use of Laboratory Animals. Figure 1A shows the overview of the study, including the cell culture models, viability, migration and immunosorbent assays, RNA sequencing, and mouse models. The study is reported in accordance with ARRIVE guidelines.
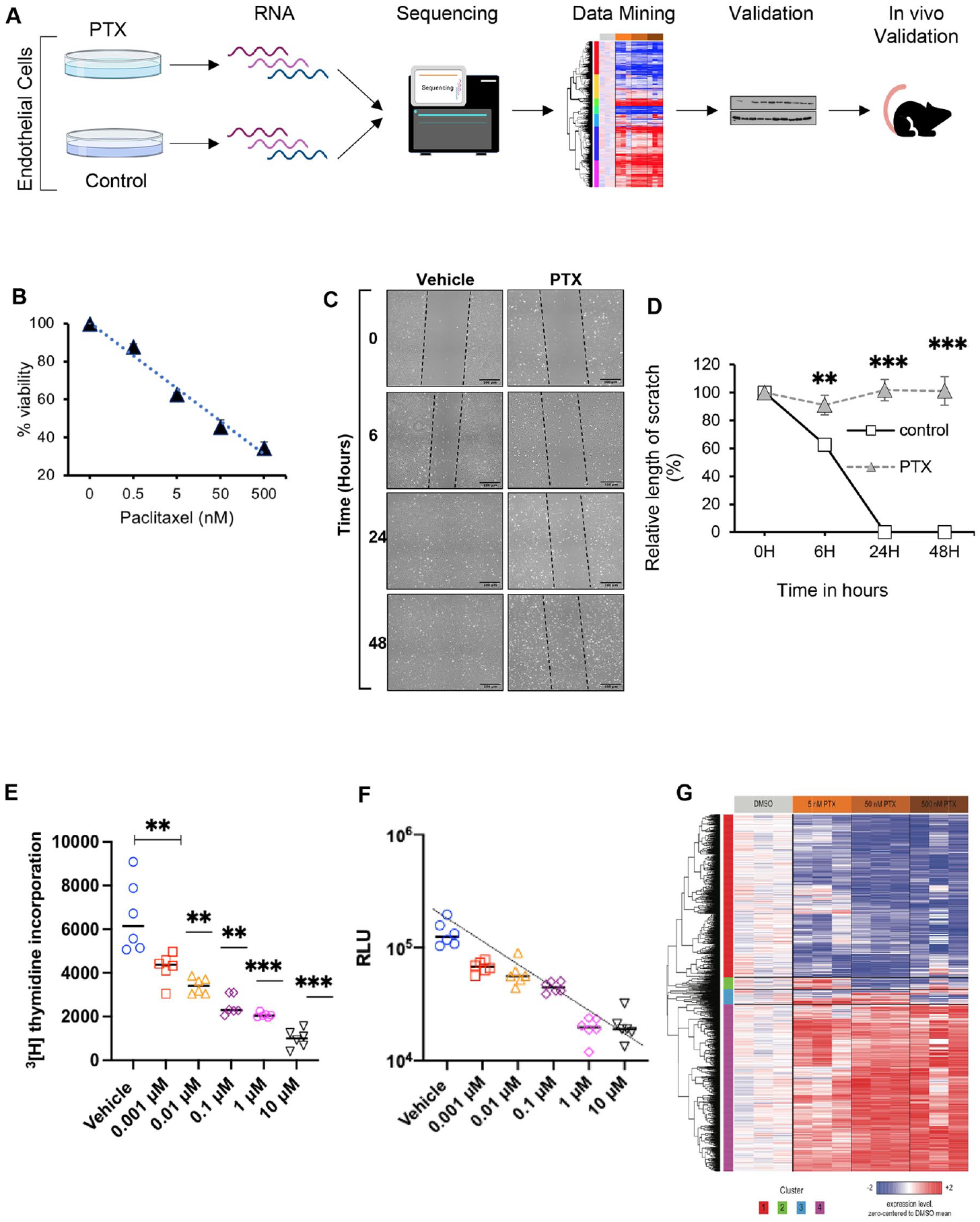
PTX) negatively inhibits endothelial cell (EC) function and induces genetic perturbation.
Cell culture
Pooled human umbilical vascular ECs (HUVECs; cat no. C2519AS) were obtained from Lonza (USA) and grown in EC growth medium (EGM)-2 media made from EC growth basal medium (EBM)-2 media supplemented with an EGM-2 BulletKit (cat no. CC-3162; Lonza). Cells were incubated in a 37°C humidified incubator with 5% CO2. ECs were grown in gelatin-coated plates. The culture plates were coated with 2% gelatin (cat no. G1890; Sigma, USA) and air dried for at least 1 h before use.
Drugs
PTX (cat no. P9600; LC Laboratories, USA) and dexamethasone (DEX) (cat no. 11015; Cayman Chemical Company, USA) were dissolved in dimethyl sulfoxide (DMSO) (cat no. D8418-250ML; Sigma).
Cell viability assay
ECs seeded in 96-well plates underwent survival assay using alamarBlue solution (cat no. BUF012B; Bio-Rad, USA) per the manufacturer’s instructions.
RNA extraction
ECs were seeded in P60 plates, treated with DMSO and PTX at ~60% confluence for 24 h, and harvested for RNA extraction following the RNeasy Mini Kit protocol (cat no. 74104; Qiagen, USA). RNA was eluted with 40 µL of RNase-free water. The RNA concentration was measured with a BioDrop spectrophotometer (Biochrom Ltd., USA).
Antibodies
Primary antibodies used were CKS2 (cat no. ab155078; Abcam, USA), BMF (Bcl-2-Modifying Factor; cat no. 50542S; Cell Signaling, USA), CD137 (cat no. 34594S; Cell Signaling), MCP-1 (cat no. 81559S; Cell Signaling), and HSP90 (cat no. 8165S; Cell Signaling). Proteins were visualized with chemiluminescent substrate (cat no. 34577; Thermo Scientific, USA) and a developing machine (Bio-Rad, USA).
Migration assay and imaging
EC cells were grown in six-well plates until 60% confluency and treated with drugs for 18 h. A single scratch with a P1000 pipette tip was induced on the monolayer. Media was aspirated and 2 mL of new drug-treated media was added after the scratch. Images were taken at baseline, immediately after the scratch, and at 6 h, 24 h, and 48 h postscratch.
Thymidine incorporation assay
A total of 10,000 primary HUVECs seeded in a 96-well plate were serum starved overnight. The cells were stimulated using EGM medium containing 5% serum for 24 h, after which they were subjected to 1 μCi of 3 H-thymidine (PerkinElmer, USA) overnight. The lysed cells were counted for radioactivity using the LabLogic 300SL Liquid Scintillation Counter (Sheffield, UK).
Apoptosis assay
An ELISA kit from Promega (cat no. 4685-096-K, USA) was used according to manufacturer instructions to measure EC apoptosis.
Image processing
All images were taken with a Nikon Eclipse Ti (USA) inverted microscope with bright-field camera at the Boston University School of Medicine (BUSM) Imaging Core. NIS-Elements software was the platform used for controlling the microscope and documenting photos. Six fields of 10× images were taken randomly with phase 1 lenses to create a wide-field image. X, Y, and Z coordinates of the scratch in each well were saved onto the microscopy software so that each time point would have the image at the same location to reduce variability. Images were exported as full-resolution tagged image file format (TIFF) and quantified with ImageJ software. The ‘straight line’ tool was used to make a random horizontal straight line across the scratch to measure the distance of the gap in three different locations of the same sample. Each sample was repeated in triplicate. The distance of the gap was taken at a random location for the same well at different times. The straight line was then added to the ‘ROI manager’ and values were measured in pixels.
Animal model
A group of female C57BL/6, 8–10-week-old mice were obtained from The Jackson Laboratory (USA) and housed by Boston University Medical Center with approval from the Institutional Animal Care and Use Committee (IACUC AN-1549). DEX and PTX were dissolved in DMSO and further diluted in normal saline for intraperitoneal (IP) injections at 5 mg/kg. Isoflurane inhalation was used as an anesthetic agent. Specifically, we used 2% inhalation for induction and 1% isoflurane inhalation for maintenance using a nose cone. Euthanasia was induced via CO2 inhalation in a precharged chamber followed by secondary euthanasia by thoracotomy. These methods are consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. Serum, heart, kidney, liver, and aorta were collected at either 20 min or 12 h after IP injection. Serum underwent ELISA and liquid chromatography/mass spectrometry (LC/MS) studies to examine various inflammatory cytokine levels and serum PTX levels. Mouse aortas were subjected to immunofluorescence staining and protein analysis.
Cytokine ELISA panel
ECs plated in six-well plates were treated with vehicle, PTX, DEX, or PTX+DEX. Next, 300 µL of media was collected and sent to Quansys Biosciences (Logan, UT, USA) to perform the Human Cytokine Release Syndrome (16-plex) ELISA panel. A total of 150 µL of mouse serum was collected and sent to Quansys Biosciences to perform the Mouse Cytokine (6-plex) ELISA panel.
Other methods, including immunoblotting, antibodies, immunohistochemistry, immunofluorescence, and LC/MS are included in the online supplementary material.
RNA sequencing and analysis
STAR (version 2.6.0c) was used to align sequence reads to human genome build hg38. FASTQ quality was assessed using FastQC (version 0.11.7), and alignment quality was assessed using RSeQC (version 3.0.0). Ensembl-Gene-level counts for nonmitochondrial genes were generated using featureCounts (Subread package, version 1.6.2) and Ensembl annotation build 100 (uniquely aligned proper pairs, same strand). Differential expression was assessed using the likelihood ratio test implemented in the DESeq2 R package (version 1.23.10) to perform a one-way analysis of variance (ANOVA) with respect to PTX concentration. Benjamini–Hochberg False Discovery Rate (FDR) correction was applied to obtain FDR-corrected p-values (i.e., FDR q-values). The web-based tool Enrichr (https://maayanlab.cloud/Enrichr/) was used to identify pathways and processes included in the MSigDB Hallmark 2020 collection that are significantly overrepresented within clusters of genes.
Statistical analyses
Parameters were expressed as mean, median, SD, and standard error of the mean (SEM). A comparison of groups was performed using independent Student’s t-tests where a p-value less than 0.05 was considered significant. In selected cases, adjustments were performed for multiple comparisons using Bonferroni correction. All p-values less than 0.05 were deemed significant as statistical analyses were performed using Excel and Prism software.
Results
Paxlitaxel (PTX) compromises endothelial cell (EC) functions and induces pro-inflammatory genes
We first examined the effect of PTX in primary HUVECs. ECs were treated with PTX for 18 h and DMSO-treated cells served as controls. PTX significantly reduced cell viability in a dose-dependent manner, reaching half-maximal inhibitory concentration (IC50) at 50 nM (Figure 1B). The effect of PTX on EC migration was examined using a scratch assay, where a monolayer of ECs was injured following treatment with PTX for 18 h (Figures 1C and 1D). The scar completely closed in the control group but the scar in the PTX-treated ECs remained unaltered over 48 h, suggesting that PTX significantly suppresses EC viability and migration. Scratch assay examines two distinct functions of ECs – proliferation and migration to heal the wound. We specifically examined proliferation using a 3 H-thymidine incorporation assay, which estimates incorporation of thymidine nucleotide during the M phase of the cell cycle. Briefly, 104 early passage primary HUVECs seeded in 96-well plates were synchronized overnight by serum starvation and stimulated by the full EGM with and without PTX. Next, 1 μCi of 3 H-thymidine (PerkinElmer) was added to each well overnight, and cells were harvested after 16–24 h of treatment. Our results showed that PTX significantly inhibited 3 H-thymidine incorporation. Compared to vehicle-treated cells, PTX 0.001 mM suppressed proliferation by 34% (p = 0.008), PTX 0.01 mM by 54% (p = 0.002), PTX 0.001 mM by 62% (p = 0.001) and more than 80% at PTX > 1 mM (p < 0.001). The IC50 of PTX is 32 nM to inhibit the incorporation of radioactive thymidine (Figure 1E).
Lastly, we posited that PTX may increase EC apoptosis, which was examined with a poly (ADP-ribose) polymerase (PARP) apoptosis assay (Figure 1F) using a chemiluminescence assay. This assay examines the induction of programmed cell death in ECs by detecting the generation of PARP. PARP-1 triggers the translocation of the apoptosis-inducing factor from the mitochondria to the nucleus and triggers the programmed cell death. This assay detects PAR deposited by PARP-1 in the EC lysates onto immobilized histone proteins of EC lysates in a 96-well format. Our data showed that paclitaxel at 4 nM concentration was sufficient to activate apoptosis. These findings suggest specific effects of PTX on fundamental endothelial functions such as proliferation and apoptosis, which, in turn, influence the survival of ECs.
RNA sequencing was then performed using ECs treated with PTX over a range of concentrations centered on its IC50 (5, 50, and 500 nM). A one-way ANOVA was used to identify genes that are differentially regulated across PTX concentrations, and the 1766 genes with the greatest significance (FDR q < 0.25) were divided into four groups by hierarchical clustering (Figure 1G
Dexamethasone (DEX) abrogates transcriptional perturbations modulated by PTX in ECs
Next, we validated specific transcriptional perturbations at the protein level by treating ECs with titrated concentrations of PTX or DEX, with the hypothesis that DEX will revert changes in expression induced by PTX. Treatment with concentrations of PTX as low as 5 nM increased CD137 expression 2.5-fold (p < 0.01) (Figures 2A and 2B). DEX treatment alone had a minimal effect on CD137 levels, except for a marginal downregulation of CD137 at 100 µM DEX (p < 0.05); however, co-treatment with DEX prevented the induction of CD137 expression by PTX (Figures 2C and 2D). Similarly, PTX up-regulated CKS2 expression in ECs in a dose-dependent manner, which was prevented by co-treatment with DEX (Figures 2E–2H). Immunoblot analysis also confirmed the observation by RNAseq that BMF expression is down-regulated by PTX (online supplementary Figure 3) and showed that it was up-regulated in a dose-dependent manner by DEX.
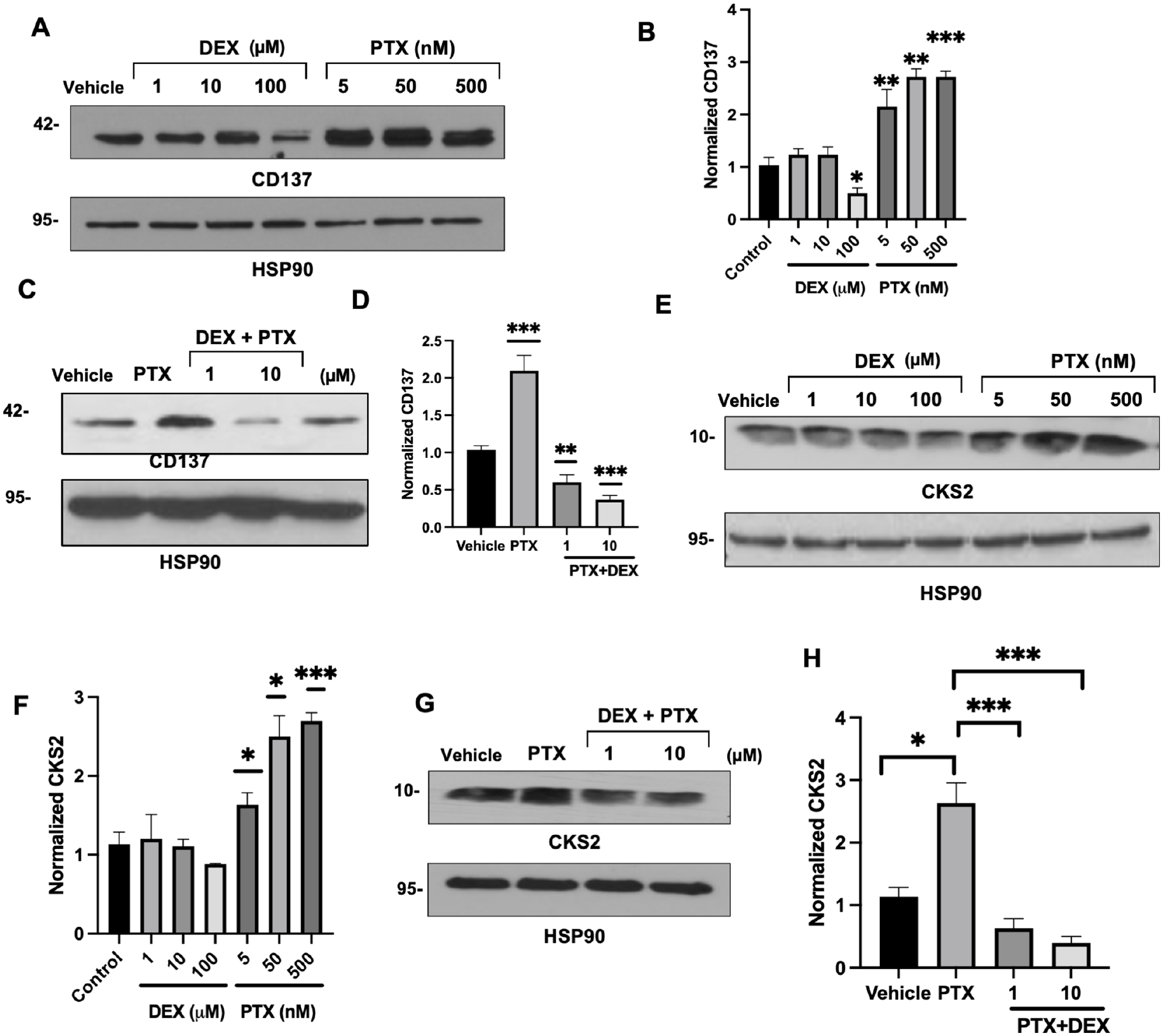
Regulation of CD137 and CKS2 protein expression in endothelial cells (ECs) by paclitaxel (PTX) and DEX.
DEX prevents the induction of MCP-1 by PTX in vitro
The RNAseq analysis showed that the transcription of MCP-1 (CCL2) is up-regulated by up to twofold in ECs by PTX in a dose-dependent manner. Accordingly, treatment with 5-nM PTX doubled the level of MCP-1 protein in ECs (Figures 3A–3D). Furthermore, co-treatment of DEX suppressed this PTX-mediated upregulation of MCP-1 in the EC lysates. MCP-1 is a secreted protein and was measured in the media of ECs using multiplex cytokine analysis. Conditioned media obtained from ECs pretreated with 5 nM or 50 nM PTX showed a significant increase in MCP-1 levels compared to vehicle-treated EC (Figures 3E–3G). Interestingly, PTX treatment up-regulated a host of pro-inflammatory cytokines in the media of ECs, including interferon (IFN)-α and IL-6, which were down-regulated by DEX in a dose-dependent manner. Collectively, these results validated transcriptional perturbations induced by PTX in ECs and supported further in vivo examination of DEX.
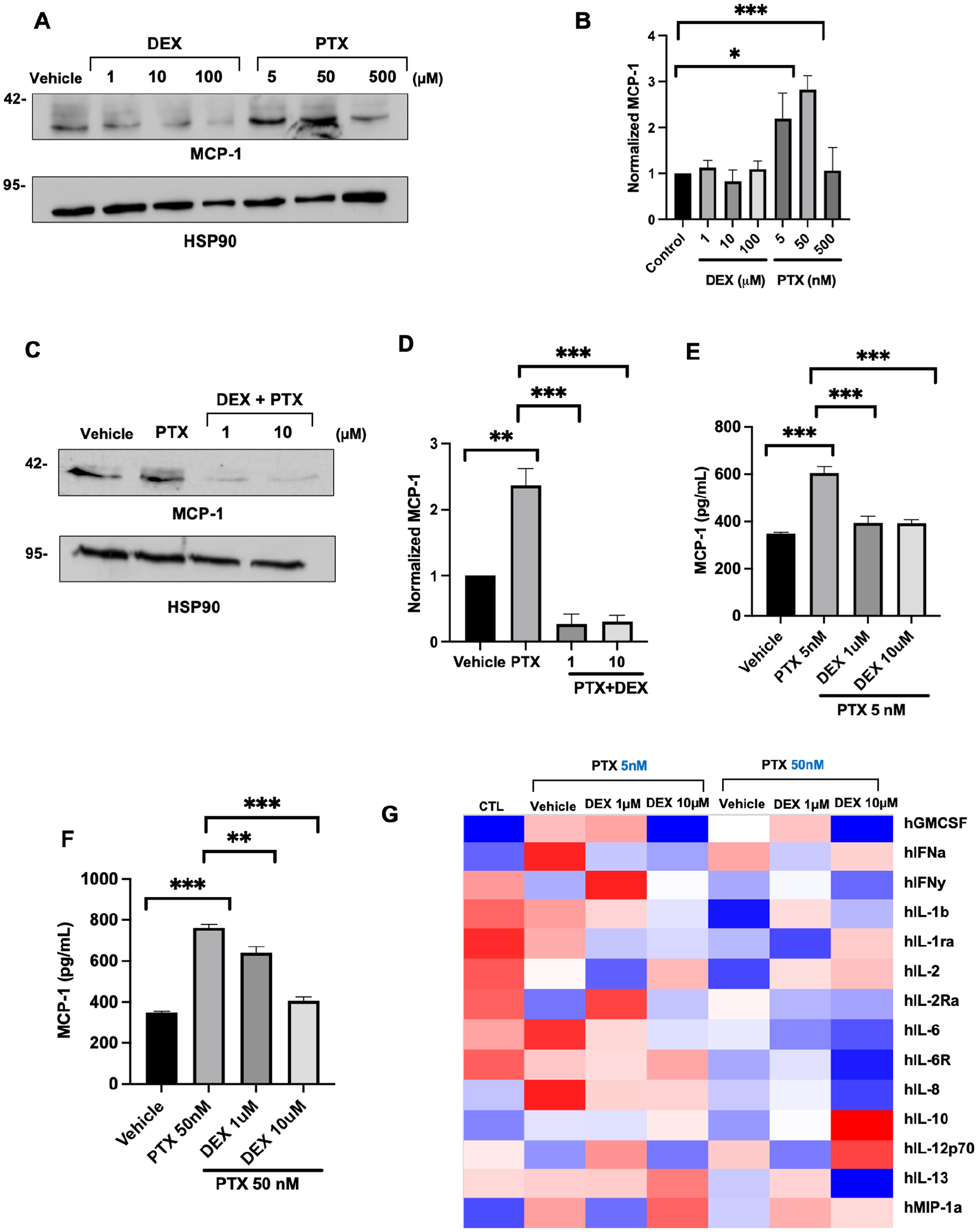
Regulation of MCP-1 and secreted inflammatory cytokines in endothelial cells (ECs) by paclitaxel (PTX) and DEX.
Peri-procedural treatment with DEX suppresses PTX-induced increase in MCP-1 levels
A group of C57BL/6 mice were randomized into four groups and administered 5 mg/kg PTX, 5 mg/kg DEX, or PTX+DEX IP. DMSO (vehicle) treated mice served as controls. Animals were harvested at either 20 min or 12 h (Figure 4A) to examine the immediate and delayed effects of PTX exposure. Serum levels of PTX were high in animals treated with PTX (average ± SEM of 6.8 ± 1.93 mmol/L) or PTX+DEX (4.99 ± 1.08 mmol/L). There was no significant difference in the PTX levels between these groups, and PTX was undetectable after 12 h. Within 20 min of PTX injection, no significant increase in MCP-1 was detected; however, by 12 h, PTX-injected mice showed a threefold increase in the levels of MCP-1 (p < 0.001) compared to vehicle-treated mice. This effect was entirely abrogated in mice treated with PTX+DEX (p < 0.001) (Figure 4B).
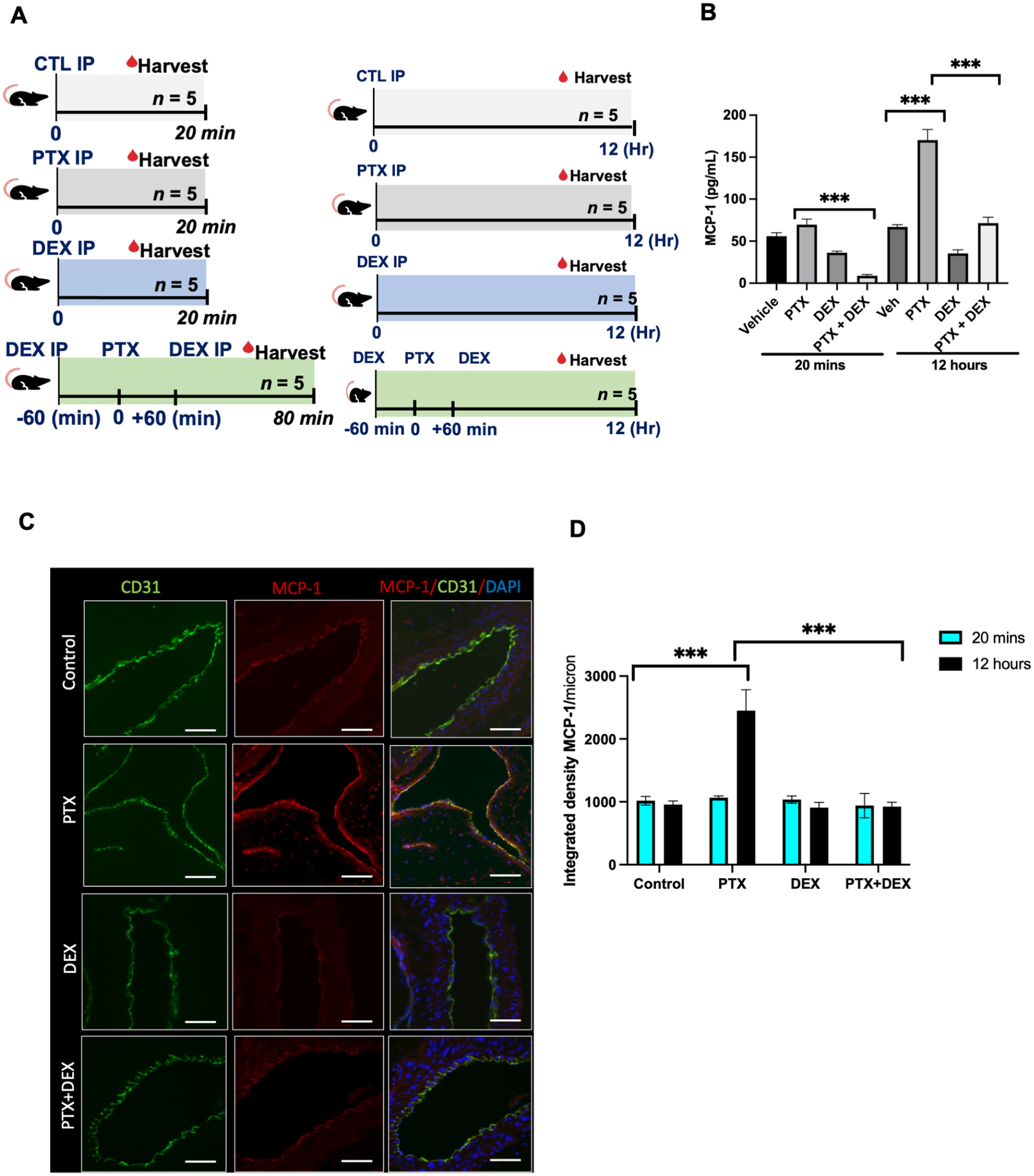
Paclitaxel (PTX) induces upregulation of MCP-1 in a mouse model.
We next examined whether PTX treatment altered MCP-1 levels in the aortic ECs of mice. The aorta of mice from different groups were stained and whole-slide imaging was subjected to ImageJ analysis to quantitate the expression of protein using an integrated density (a composite of image intensity and the number of pixels normalized to the area). CD31 was used as an EC marker. There were no significant changes in MCP-1 expression at 20 min (online supplementary Figure 4). However, at 12 h, PTX-exposed mice showed a 2.3-fold (p < 0.001) increase in MCP-1 expression compared to control mice (Figures 4C–4D), which was completely suppressed in the mice co-treated with PTX+DEX (p < 0.001). Similarly, no significant changes in CD137 levels were noted within 20 min of PTX treatment (online supplementary Figure 5), but at 12 h a significant upregulation of CD137 was noted in the aortic ECs of PTX-treated mice (p < 0.05), which was absent in mice injected with PTX+DEX (p < 0.001) (Figures 5A and 5B).
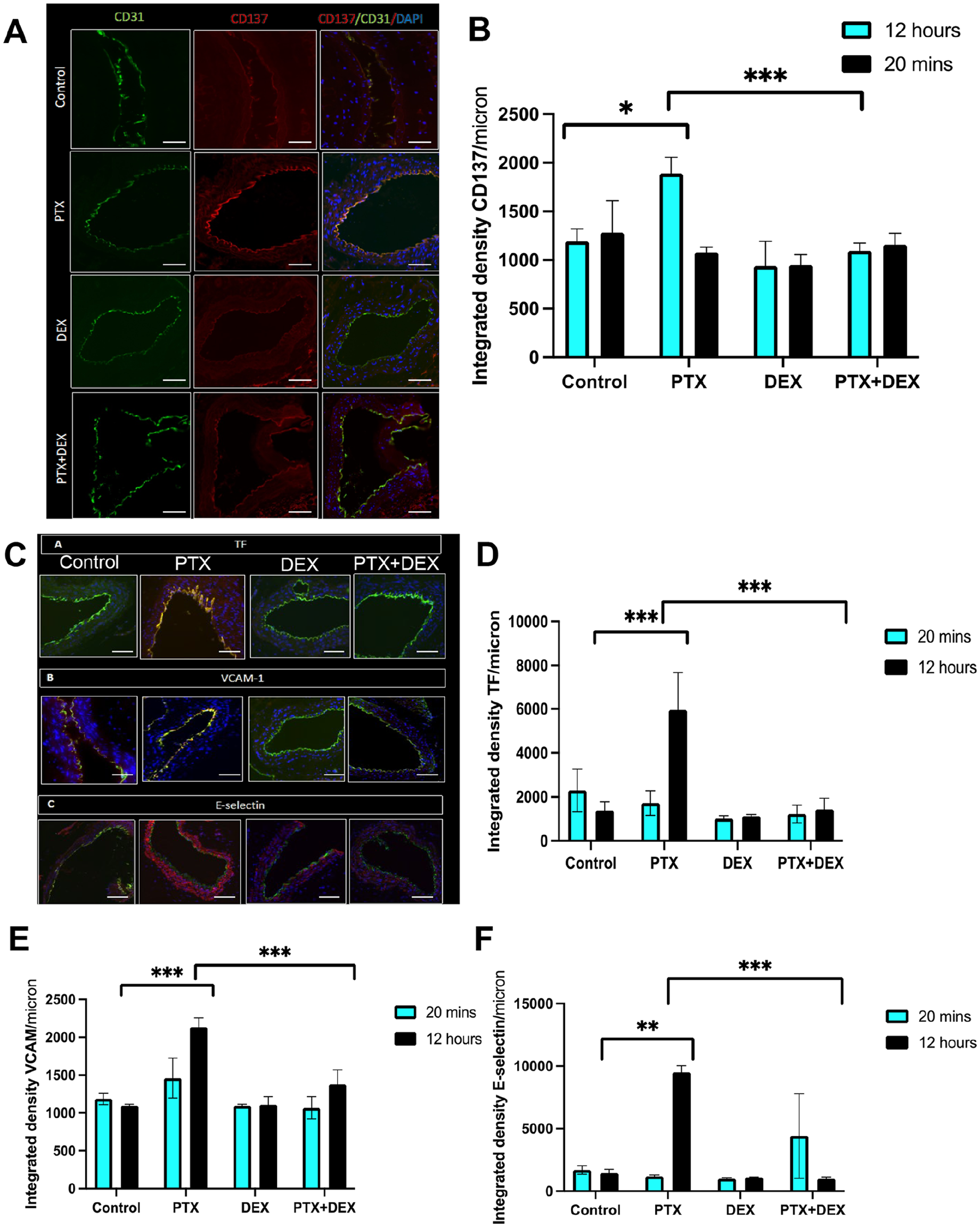
Paclitaxel (PTX) induces expression of pro-inflammatory and pro-thrombotic proteins in mouse aortic endothelial cells.
DEX suppresses the induction of pro-thrombotic mediators by PTX
Pro-inflammatory mediators such as MCP-1 and CD137 induce atherothrombotic factors through downstream mediators such as tissue factor (TF), E-selectin, and VCAM-1, converting normal vascular endothelium from an anticoagulant to a pro-coagulant state.15,16 TF is the primary trigger of the extrinsic coagulation cascade, and its upregulation increases the risk of plaque rupture and cardiovascular events. We therefore evaluated whether PTX increased the expression of these downstream prothrombotic mediators in ECs (Figure 5C).
At 20 min after PTX injection, there was no significant change in TF levels between the PTX and control groups (online supplementary Figure 6). However, by 12 h, TF expression was significantly increased in the PTX group compared to controls (p < 0.001), and this increase was abrogated by co-administration of PTX+DEX (p < 0.001) (Figures 5C and 5D). A similar pattern was observed with respect to VCAM-1 and E-selectin expression detected by immunofluorescence assay and Western blots (Figures 5E and 5F; online supplementary Figures 7–9).
Discussion
We show that a single systemic exposure of PTX in mice is sufficient to upregulate pro-inflammatory proteins and atherothrombotic mediators, which are likely to trigger subsequent events leading to vascular toxicity. Our in vivo experimental strategy probed two time points – 20 min and 12 h postinjection – to evaluate the immediate and delayed effects of PTX exposure. Within 20 min after PTX injection, both the PTX and PTX-DEX-treated groups achieved comparable PTX blood levels (4.99–6.88 mmol/L) which are similar to concentrations noted in previous studies.17,18 Considering we identified the IC50 of PTX as 50 nM for ECs in vitro, this systemic concentration implies that ECs in vivo were subjected to toxic drug levels.
MCP-1 or CCL2 is one of the key chemokines of the CC family, which regulates migration and recruitment of monocytes and increases cytokine production, adhesion molecule expression, and induction of reactive oxygen species (ROS) release in monocytes and ECs. 19 MCP-1 is expressed by a variety of activated cells (e.g., ECs, monocytes, and smooth muscle cells). 19 Though our work focused on ECs as a source of MCP-1 in blood, it does not rule out the possibility of an increase in MCP-1 from other cell types. Using genetic models, several studies have demonstrated the importance of MCP-1 in the development of atheromatous lesions and its consequences. For example, higher MCP-1 levels were detected in atherosclerotic lesions compared to normal human arteries.20,21 Rupture of an atherosclerotic plaque is known to be associated with thrombotic events with potentially life-threatening complications. Studies have also demonstrated the proangiogenic effects of MCP-1 in the atherosclerotic plaque, which is known to increase plaque vulnerability. 22 MCP-1 induces matrix metalloproteinases leading to thinning of the atherosclerotic cap and increasing the risk of plaque rupture. 23 In addition, MCP-1 upregulates TF synthesis, further contributing to thrombosis on the ruptured plaque and increasing the risk of potentially fatal coronary events. 15 Considering these known effects of MCP-1, the current data raise a possibility to relate the perceived long-term effect due to high levels of MCP-1 following PTX exposure, and this may likely contribute to the initiation of atherothrombotic processes.
Upregulation of CD137 in ECs leads to endothelial dysfunction, which subsequently increases the expression of adhesion molecules on ECs and pro-inflammatory cytokine production by them; both of these processes augment the recruitment and migration of leukocytes to exacerbate the atherosclerotic process.13,14 Based on the above findings, it is conceivable that a high dose of systemic exposure of PTX upregulates mediators that are known to trigger cascades of secondary events increasing the atherothrombotic process in the vasculature.
DEX is a synthetic glucocorticoid that exerts profound antiinflammatory effects via different mechanisms, including platelet-derived growth factor (PDGF) and IL-1β inhibition. 24 In addition to suppressing cytokine and interleukin signaling, DEX is known to reduce vascular smooth muscle cell migration and proliferation and to downregulate adhesion molecule expression on ECs at the site of vessel injury, 24 all of which constitute the rationale of using DEX in this study. Our selection of DEX was further motivated by its ease of administration in the clinical setup before and after endovascular interventions.
Study limitations
Our study has a few limitations. We used cell culture and animal models to demonstrate the antiinflammatory effect of DEX after PTX exposure. PTX dosing in our experiments was based on the overall drug content on a prototypic 200 mm length DCB approved for human use (RangerTM; Boston Scientific; Marlborough, MA, USA) and also considering the systemic exposure of patients undergoing chemotherapy. Future studies can explore varied levels of PTX dosing followed by evaluation of systemic effects. Furthermore, detailed human studies are needed to assess the blood concentration of PTX during the deployment of PTX devices, which can inform animal experiments. In this work, we utilized HSP90 as a loading control for immunoblotting due to its differing molecular weight from our proteins of interest. However, we acknowledge that the variable expression of HSP90 in various pathological conditions poses a limitation to this approach. Studies are also needed to rigorously evaluate the effect of PTX over a protracted period in higher models such as swine and the ones with comorbidities commonly seen in patients with high cardiovascular disease burden (such as obesity, diabetes, etc.). Large animal studies are also needed to evaluate systemic toxicity when drug release is simulated following DCB angioplasty and to account for biophysical and biochemical interactions.
Conclusion
In conclusion, this work demonstrates potential mediators of PTX-induced systemic toxicity emanating from a high-dose exposure. This spike likely contributes to systemic toxicity, thereby enhancing the risk of atherothrombotic progression. The strategy to minimize PTX-induced toxic effects by systemically administering widely used antiinflammatory compounds such as DEX should be explored further using other preclinical models and clinical studies. We hope that this study paves the way for further analysis to broadly understand the mechanisms and mediators of PTX-induced adverse cardiovascular events observed in humans and the means to prevent them.
Supplemental Material
sj-docx-2-vmj-10.1177_1358863X241231942 – Supplemental material for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment
Supplemental material, sj-docx-2-vmj-10.1177_1358863X241231942 for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment by Mengwei Zhang, Saran Lotfollahzadeh, Nagla Elzinad, Xiaosheng Yang, Murad Elsadawi, Adam C Gower, Mostafa Belghasem, Tarek Shazly, Vijaya B Kolachalama and Vipul C Chitalia in Vascular Medicine
Supplemental Material
sj-docx-3-vmj-10.1177_1358863X241231942 – Supplemental material for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment
Supplemental material, sj-docx-3-vmj-10.1177_1358863X241231942 for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment by Mengwei Zhang, Saran Lotfollahzadeh, Nagla Elzinad, Xiaosheng Yang, Murad Elsadawi, Adam C Gower, Mostafa Belghasem, Tarek Shazly, Vijaya B Kolachalama and Vipul C Chitalia in Vascular Medicine
Supplemental Material
sj-docx-4-vmj-10.1177_1358863X241231942 – Supplemental material for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment
Supplemental material, sj-docx-4-vmj-10.1177_1358863X241231942 for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment by Mengwei Zhang, Saran Lotfollahzadeh, Nagla Elzinad, Xiaosheng Yang, Murad Elsadawi, Adam C Gower, Mostafa Belghasem, Tarek Shazly, Vijaya B Kolachalama and Vipul C Chitalia in Vascular Medicine
Supplemental Material
sj-pdf-1-vmj-10.1177_1358863X241231942 – Supplemental material for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment
Supplemental material, sj-pdf-1-vmj-10.1177_1358863X241231942 for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment by Mengwei Zhang, Saran Lotfollahzadeh, Nagla Elzinad, Xiaosheng Yang, Murad Elsadawi, Adam C Gower, Mostafa Belghasem, Tarek Shazly, Vijaya B Kolachalama and Vipul C Chitalia in Vascular Medicine
Supplemental Material
sj-xlsx-5-vmj-10.1177_1358863X241231942 – Supplemental material for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment
Supplemental material, sj-xlsx-5-vmj-10.1177_1358863X241231942 for Alleviating iatrogenic effects of paclitaxel via antiinflammatory treatment by Mengwei Zhang, Saran Lotfollahzadeh, Nagla Elzinad, Xiaosheng Yang, Murad Elsadawi, Adam C Gower, Mostafa Belghasem, Tarek Shazly, Vijaya B Kolachalama and Vipul C Chitalia in Vascular Medicine
Footnotes
Acknowledgements
We thank Dr Michael Kirber at the BUMC Imaging Core facility for his assistance in confocal microscopy.
Availability of data and material
Declaration of conflicting interest
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was funded by grants from the National Institutes of Health (R01-HL159620, R21-DK119740, R21-CA253498, RF1-AG-062109, R21 DK119740, HL-166608, and UL1TR001430, 1R01HL166608, R21 DK119740), and the American Heart Association (17SDG33670323, 20SFRN35460031, and CAT-HD Center grant # 857078), and Center of Cross-Organ Vascular Pathology (Boston University).
Supplementary material
The supplementary material is available online with the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.