
Abstract
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease, is a rare disorder with a case prevalence as high as one in 5000, causing arteriovenous malformations in multiple organ systems. HHT is familial with autosomal dominant inheritance, with genetic testing allowing confirmation of the diagnosis in asymptomatic kindreds. Common clinical manifestations are epistaxis and intestinal lesions causing anemia and requiring transfusions. Pulmonary vascular malformations predispose to ischemic stroke and brain abscess and may cause dyspnea and cardiac failure. Brain vascular malformations can cause hemorrhagic stroke and seizures. Rarely, liver arteriovenous malformations can cause hepatic failure. A form of HHT can cause juvenile polyposis syndrome and colon cancer. Specialists in multiple fields may be called to care for one or more aspects of HHT, but few are familiar with evidence-based guidelines for HHT management or see a sufficient number of patients to gain experience with the unique characteristics of the disease. Primary care physicians and specialists are often unaware of the important manifestations of HHT in multiple systems and the thresholds for their screening and appropriate management. To improve familiarity, experience, and coordinated multisystem care for patients with HHT, the Cure HHT Foundation, which advocates for patients and families with this disease, has accredited 29 centers in North America with designated specialists for the evaluation and care of patients with HHT. Team assembly and current screening and management protocols are described as a model for evidence-based, multidisciplinary care in this disease.
Keywords
Introduction
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease, is a rare disorder with a case prevalence as high as one in 5000, causing abnormal blood vessel formation.1,2 Characteristics include the development of arteriovenous malformations (AVMs) of varying severity in the oronasal mucosa, skin, lungs, brain, gut, and liver. Clinical sequelae include anemia, difficulty controlling nosebleeds, gastrointestinal (GI) bleeding, and resulting anemia that requires frequent transfusions and other treatment. Pulmonary AVMs (PAVMs) can cause embolic stroke, brain abscess, pulmonary hypertension, and other respiratory complications. Brain AVMs (BAVMs) can cause hemorrhagic stroke and seizures. In rare cases, AVM of the liver can cause varying degrees of liver failure. Cases are inherited in an autosomal dominant fashion, with mutations at three known loci and high clinical penetrance (> 80% of those affected harbor at least one characteristic requiring clinical intervention).1–5 Overall, HHT and associated comorbidities may impact life expectancy, 6 and there is a huge opportunity for proactive management of many HHT-related complications.
Evidence-based guidelines for the management of this disease have been published, but much of the level of evidence is weak.1–4,7 Specialists in multiple fields may be called to care for one or more aspects of HHT, but few are familiar with these guidelines or see a sufficient number of patients to gain experience with the various disease characteristics. Primary care physicians and specialists are often unaware of the manifestations of disease in multiple systems and the thresholds for their screening and appropriate management. To improve familiarity, experience, and coordinated multisystem care for patients with HHT, the Cure HHT Foundation (curehht.org), which advocates for patients and families with this disease, has accredited 29 centers in North America with designated specialists for the evaluation and care of patients with HHT. 8 These centers and the Cure HHT Foundation website serve as resources to community physicians and other specialists. However, to date, there has not been a publication on how such a center is organized and how screening and multidisciplinary care of HHT are effectively implemented. The spectrum of disease manifestations in clinical practice are reviewed here, and practical recommendations are provided on the implementation of specific evidence-based care protocols related to HHT in different organ systems.
Diagnosis and disease genetics
A definitive diagnosis of HHT can be established based on clinical criteria, or by genetic testing demonstrating a pathogenetic mutation associated with the disease (Table 1). The presence of three or more clinical manifestations known as the Curaçao criteria, 7 named after the Netherlands Antilles island where the disease is endemic, establish the diagnosis of HHT. These criteria are: recurrent epistaxis, multiple telangiectasia of skin, lip or tongue (Figure 1), arteriovenous malformations of one or more internal organ, and family history of these manifestations. There is a possibility of HHT when less than three Curaçao criteria are met.
Diagnosis of hereditary hemorrhagic telangiectasia (HHT).

Note: 97% of cases with clinical diagnostic criteria carry a pathogenetic mutation at one of the three known loci; more than 80% of mutation carriers manifest clinical features of the disease and pass it to their offspring by autosomal dominant inheritance.
Cases with HHT3/SMADA4 mutations manifest juvenile polyposis syndrome, and are vulnerable to colon cancer, in addition to HHT clinical manifestations (‘Curaçao criteria’).
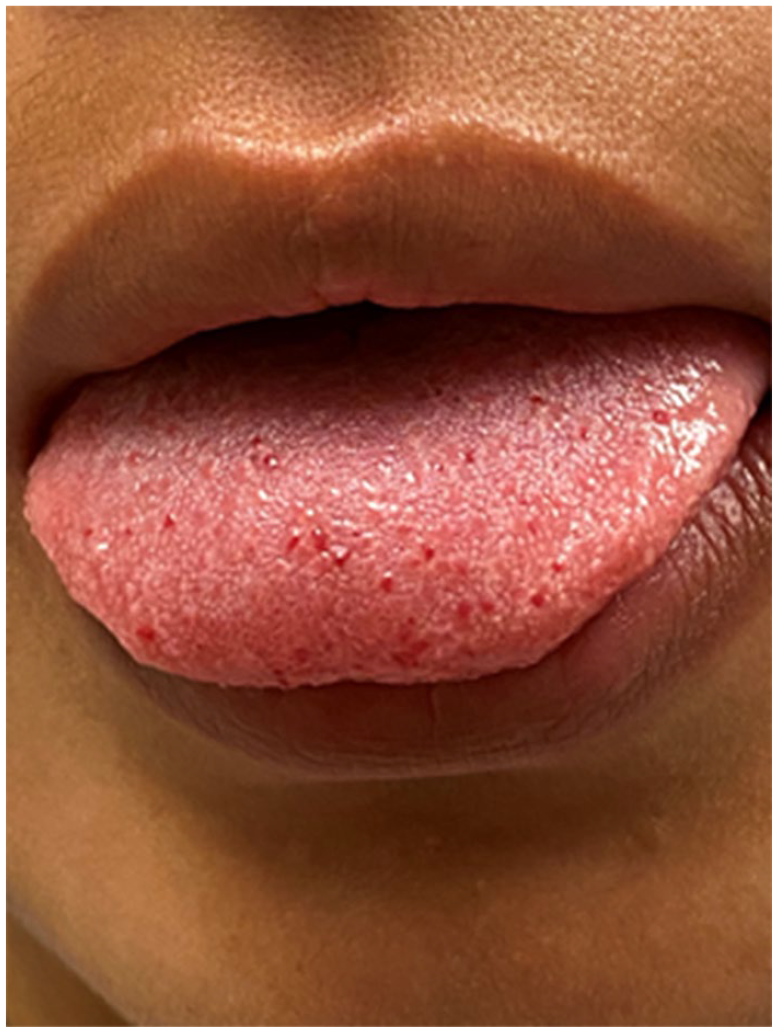
Tongue telangiectasias in a patient with HHT. These and similar lesions on the lips and fingertips contribute to clinical diagnosis. The punctate lesions are in fact micro-AVMs and rarely can cause bleeding and require treatment.
Genetic testing can clarify the diagnosis in patients who do not meet clinical criteria and can diagnose or exclude HHT in asymptomatic at-risk family members. 9 Clinical testing is available in multiple laboratories and is usually done by panel testing of genes causative of HHT. The initial testing of newly diagnosed families should start with an individual who meets the clinical criteria. 7 Pathogenetic mutations of ENG, ACVRL1, and SMAD4 account for 52%, 44%, and 1% of affected cases, respectively. 10 Approximately 3% of clinically diagnosed individuals will not have a positive mutation identified due to technical limitations of the testing and genes that are yet to be discovered. Once the specific mutation and gene have been identified in a family, it is not necessary to test other individuals with a positive clinical diagnosis based on the Curaçao criteria as it is reasonable to assume that they carry the same familial mutation. 7 Genetic test results may show variants of unclear significance in HHT genes, and these cannot be used as the only factor to determine whether an individual has the disease. Identification of a SMAD4 mutation carries additional risks and implies additional management recommendations for juvenile polyposis (see section on gastrointestinal manifestations).
HHT is an autosomal dominant condition and there are often multiple at-risk individuals who are the offspring of an affected person and, therefore, are at a 50/50 risk. The published guidelines recommend that asymptomatic children of an affected parent should be considered to have possible HHT unless excluded by negative genetic testing for the known familial mutation. Individuals who did not inherit the familial mutation do not have HHT, and their offspring are not at risk.
Genetic testing can be complicated by several issues that impact implementation. Insurance coverage of genetic testing varies widely and testing costs can be a limitation. With large pedigrees, people who need testing may be spread over large geographic regions outside of the clinical practice of the HHT center. Despite these limitations, most families can be tested and can use genetics to inform healthcare recommendations. Other tests such as nail fold capillary microscopy do not contribute additional sensitivity or specificity beyond clinical criteria and genetic testing. 11
Multidisciplinary team organization, screening protocols, and spectrum of disease manifestations
Accredited HHT centers in North America are led by a medical director or designees who evaluate every patient and coordinate necessary multidisciplinary consultations. One or more specialists in respective organ systems affected by the disease are designated among their peers to see patients with HHT preferentially, and to educate their specialty colleagues about subtle aspects of disease management. They also serve as a source of information regarding HHT to physicians outside the institution. A suggested flow chart for evaluating cases with suspected HHT diagnosis is presented in Figure 2.
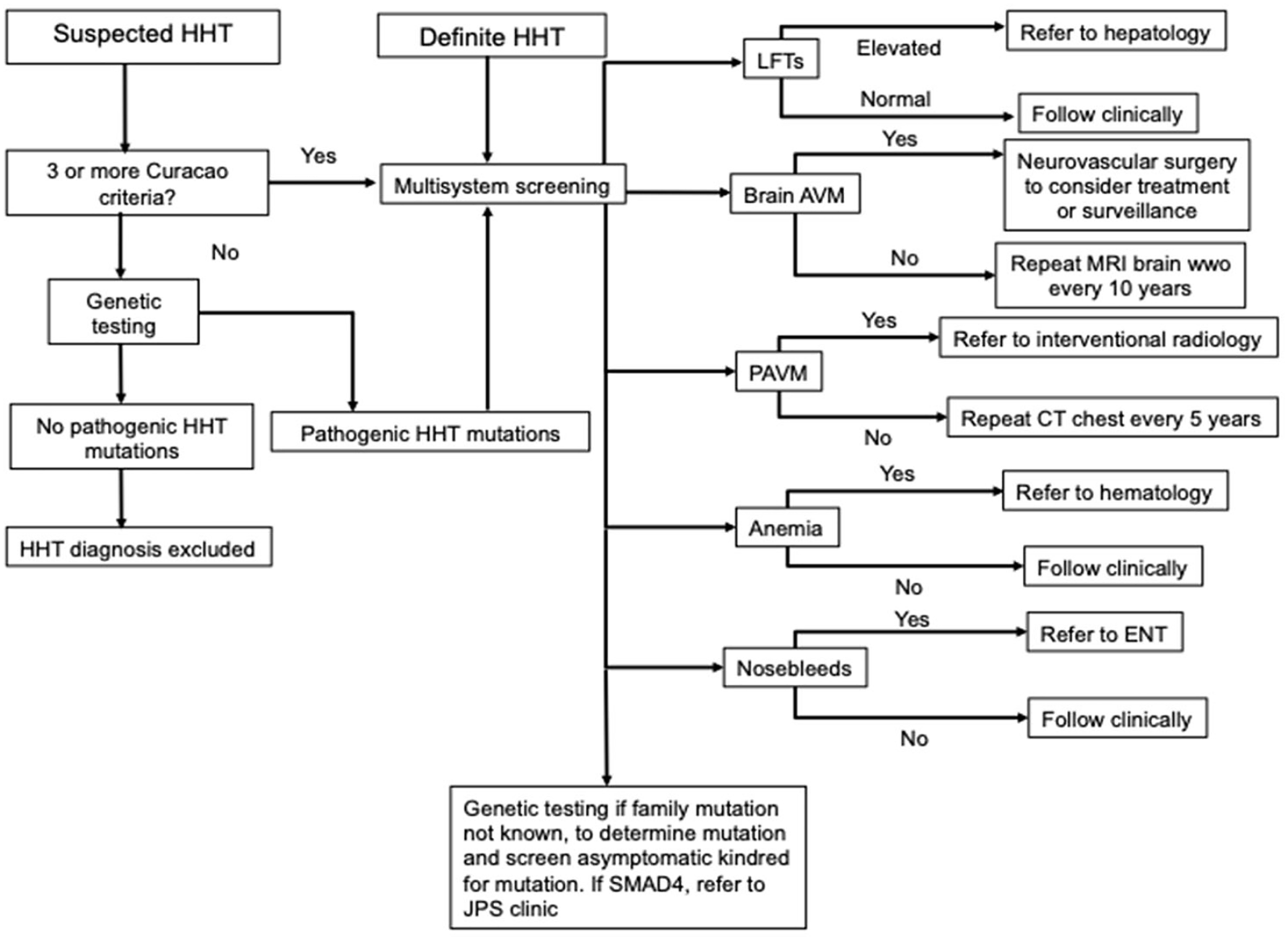
Suggested algorithm for multisystem evaluation of patients with HHT.
An example of such a multidisciplinary team at our institution includes the co-authors of this review. A designated Clinical Coordinator plays another important role, performing intake assessments of all cases with suspected HHT, and helping patients navigate multidisciplinary care (appointments, imaging, procedures, etc.). A notification tool is instituted in our institution’s electronic medical record, notifying the Program Director whenever HHT is entered as a diagnosis or clinical problem (ICD-9 448.0 / ICD-10 178.0 / ORPHA 774) on any patient at our primary and affiliated hospitals and clinics. This generates a follow-up by the HHT Clinical Coordinator with the treating team about needed screenings and evaluations. A perspective on the volume of activity is reflected by data compiled for center accreditation. A total of 236 patients were evaluated for suspected HHT between 2013 and 2022. The majority of patients are women (55%) with a mean age of 43.2. Within our cohort, 17% of cases are under 20 years of age. The clinical manifestations requiring specialist referral are noted in Figure 3, and most commonly involved nosebleeds, PAVMs, and anemia. The demonstrated genotypes include HHT1/ENG mutation, HHT2/ACVRL1 mutation, and HHT3/SMADA4 mutation in 31%, 18%, and 4% of cases, respectively. There were noncontributory genetic tests (no definite pathogenetic mutation despite clinical diagnosis of HHT) in 5% of cases. Genetic diagnosis of HHT was not confirmed in 38% of cases, including those who declined genetic testing, or their insurance did not allow it.
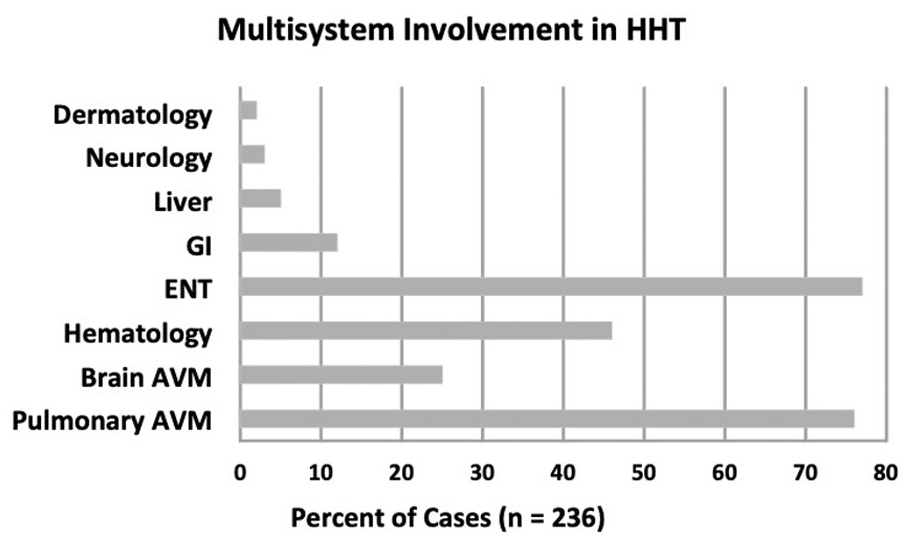
Multisystem involvement requiring specialist referral or treatment in 236 patients evaluated for suspected or definite HHT.
Special considerations in children
As in adults, the Curaçao criteria diagnose HHT in children and adolescents who meet three criteria.12,13 However, skin and oral lesions are often not present until adulthood, and even epistaxis may not manifest until the teen years.14–17 Given the frequent lack of overt symptoms and signs, current guidelines recommend that genetic tests be offered in asymptomatic children of a parent with HHT. 7
Cases with suspected or proven HHT are screened with blood work to evaluate for anemia and liver dysfunction. A saline contrast screening echocardiogram (bubble study) is performed to assess for the presence of PAVMs, and this is repeated every 5 years if negative. Large PAVMs and those associated with decreased oxygen saturation are treated with embolization. 7 At our institution, parents are given the option of screening for BAVMs with magnetic resonance imaging (MRI)/magnetic resonance angiography (MRA) at the time of diagnosis, although this may be delayed in asymptomatic infants until they can undergo brain MRI without anesthesia. An associate medical director of our HHT center oversees all clinical evaluations of children with established or suspected HHT.
Epistaxis
Designated otolaryngologists with special interest in HHT see the adult and pediatric referrals with known or suspected HHT. For the majority of cases with epistaxis, specialist input avoids potentially harmful effects of indiscriminate cauterization for nosebleeds that can stir up more bleeding from mucosal AVMs or cause perforation complications.
The management of epistaxis in patients with HHT is summarized in a flow diagram (Figure 4). It begins with aggressive nasal hygiene via moisturization to prevent bleeding caused by desiccation of vascular malformations in the anterior nasal cavity. Several products are used, including nasal saline (sprays and gels), ointments (e.g., mupirocin, petrolatum), and emollients (e.g., rose geranium oil, Ponaris). The choice of product and frequency is customized to the patient and is guided by the experience of the physician in light of direct nasal endoscopy. The most recent international guidelines suggest using topical saline spray or gel twice daily based on evidence showing reduced Epistaxis Severity Scores (ESS) with topical saline from a randomized controlled trial (RCT).7,18 Oral tranexamic acid 3 g daily (1.5 g twice daily or 1 g thrice daily) is used if nasal moisturization is unsuccessful, based on data showing a decrease in epistaxis severity from two RCTs.7,19,20 In the setting of active epistaxis, direct pressure is recommended, cold packs to the neck, and head elevation. Packing is reserved for severe or life-threatening bleeding, as placement may exacerbate future episodes due to mucosal trauma.
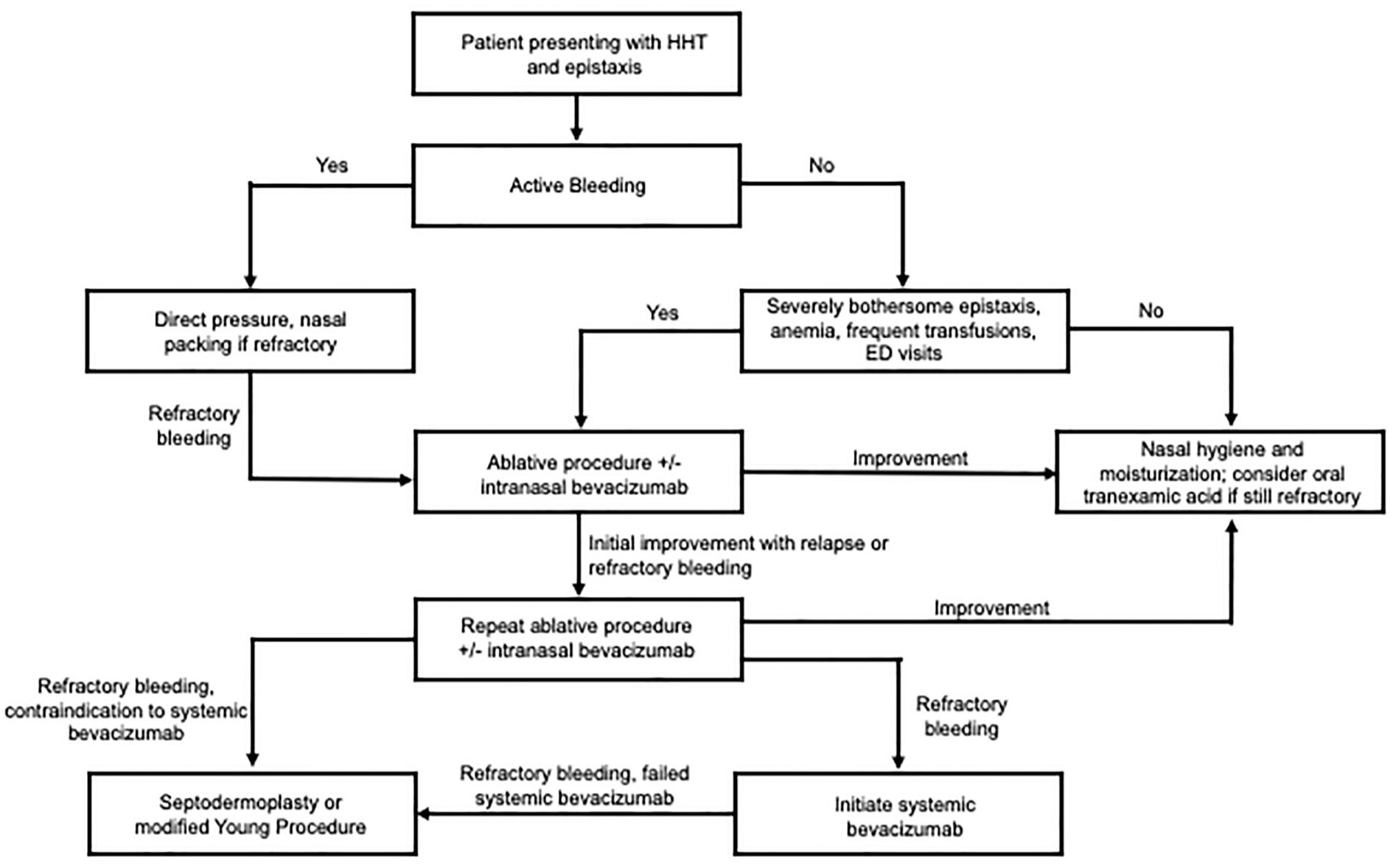
Suggested algorithm for approach to management of epistaxis in a patient with HHT.
After these conservative measures, a variety of surgical interventions may be considered. Indications include objective parameters (anemia, iron deficiency, emergency visits, packing), as well as quality of life concerns. These are evaluated informally or formally, for example, through the ESS, a validated tool to assess the severity of epistaxis by frequency and severity of episodes and medical consequences such as anemia and transfusions. 21 The type of procedure performed is customized in light of the patient’s nasal examination (Figure 5) and the severity of symptoms. Standard cauterization with monopolar and bipolar diathermy may be used for mild cases, and ablation of vascular malformations utilizing a laser or coblation device is employed for more severe disease. Sclerotherapy, most commonly with the agent sodium tetradecyl sulfate (STS), can be effective as an alternative, although this technique can have serious risks and must be implemented with caution. 22 For all patients requiring operative procedures, we also offer the option of intranasal injection of 0.5 mL of bevacizumab 25 mg/mL in divided doses in the sphenopalatine foramen, posterior bony nasal septum, upper lateral nasal wall, and nasal floor bilaterally. 23 A recent prospective, double-blind RCT comparing this protocol with bipolar cauterization alone showed a clinically significant benefit 4 months after treatment. 24
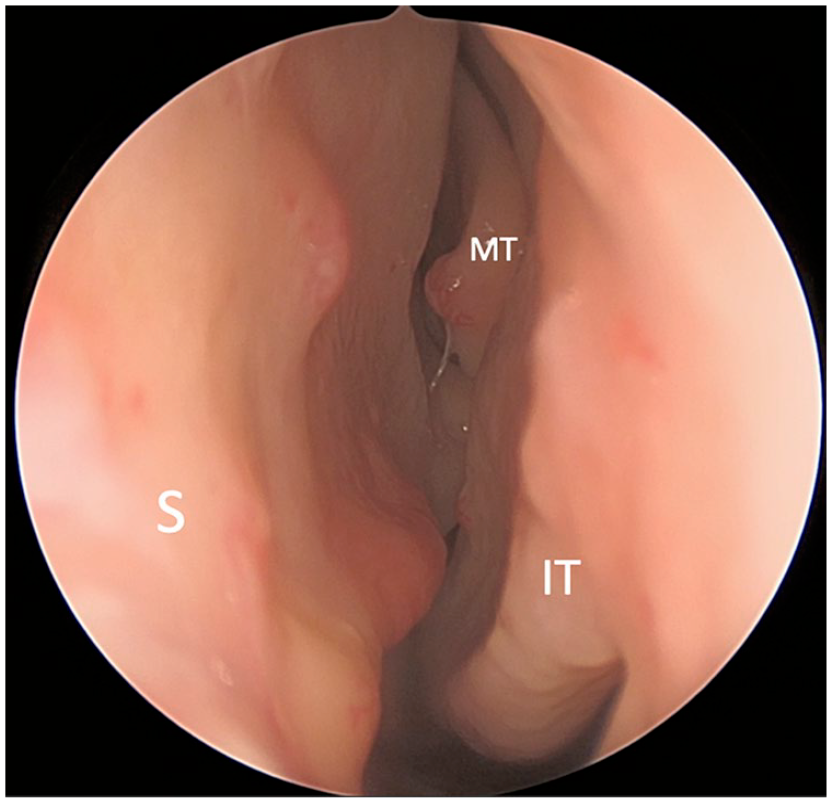
HHT endonasal examination. Left nasal cavity with multiple flat and raised telangiectasias of nasal septum (S) and one raised telangiectasia of the middle turbinate (MT) and inferior turbinate (IT). Telangiectasias are preferentially treated on the side of the nose with clinically worse epistaxis.
Alternatives for patients with severe epistaxis refractory to these measures include more invasive surgical procedures, such as septodermoplasty, which involves the removal of the respiratory epithelium of the anterior nasal septum with skin grafting and modified Young’s procedure (nasal closure). These procedures are generally avoided as they can fail, are morbid, have potentially bothersome side effects (foul nasal odor, septal perforation), or may result in other clinical compromises (significant impairment of nasal breathing, particularly when eating; nasal dryness).25–27
Systemic bevacizumab is considered in patients with persistent and severe epistaxis. A recent multicenter international observational trial of systemic bevacizumab for patients with moderate–severe HHT-associated bleeding (42% of whom were treated predominantly for epistaxis) showed significant improvement in ESS, a reduction of 82% in blood transfusions, a 70% reduction in iron transfusions, and increased hemoglobin (gain of 3.2 g/dL). 28 The authors’ experience mirrors these findings.
When a child with HHT presents with epistaxis, a careful history is obtained to assess the severity of epistaxis and the degree of interference with daily life. An epistaxis severity score is generated and previous therapies and benefits are queried. A careful examination for telangiectasias in the oral and nasal cavities is performed by anterior rhinoscopy supplemented by nasal endoscopy when tolerated. Emollients and saline humidification are instituted. If epistaxis is severe, an endoscopy in the operating room with potassium titanyl phosphate (KTP) laser ablation of telangiectasias is performed in conjunction with bevacizumab mucosal injections as necessary.
Anemia and hematologic manifestations
Hematologists have essential roles in the multidisciplinary care of patients with HHT. Owing to chronic blood loss, it is common for patients to develop iron deficiency anemia. However, patients can have other causes of anemia and other sources of bleeding that are not related to HHT. Therefore, hematologists can assist with comprehensive evaluations for other causes of anemia, as well as unrelated bleeding diathesis and coagulopathies. In terms of management, hematologists can help replace iron and consider systemic therapies to reduce bleeding and iron loss. Again, a designated hematologist can achieve greater experience and familiarity with HHT-related protocols.
Current consensus guidelines recommend that patients with HHT be monitored for iron deficiency.7,29 In our center, patients identified with iron deficiency are referred to a hematologist with experience in care of patients with HHT. Although many hematologists treat patients with iron deficiency, the ongoing and significant blood loss experienced by patients with HHT may require closer follow-up and more aggressive iron repletion. Hematologists with experience in patients with HHT can anticipate these needs, and scoring systems such as ESS can help guide management. 21 Furthermore, by regularly evaluating iron loss and bleeding, hematologists can efficiently refer patients to other specialties for the local intervention of bleeding sites. Additionally, hematologists can initiate systemic therapies to help patients maintain iron homeostasis and reduce symptomatic bleeding.
International guidelines recommend testing all adults with HHT for iron deficiency and anemia at the initial evaluation, although ongoing surveillance is individualized. 7 Ferritin measurement is quite helpful, as low ferritin levels are diagnostic of iron deficiency. 30 However, since ferritin is also an acute phase reactant, it can also be beneficial to measure total iron binding capacity and transferrin saturation. Owing to concern for continued blood loss, recommendations suggest a ferritin goal of more than 50 ng/mL. 31 At the initial evaluation, we also recommend ruling out coagulopathies with measurement of the prothrombin time and the activated partial thromboplastin time. Patients with GI AVMs can develop acquired von Willebrand syndrome. 32
Guidelines recommend that patients with iron deficiency should be treated with oral iron therapy first unless they have severe bleeding requiring more rapid correction.7,29 Oral iron can be administered every other day, and increasing dosing daily can reduce iron absorption. 33 Individual patients may benefit from and tolerate multiple daily doses. However, iron deficiency can be difficult to treat in patients with severe and ongoing bleeding with oral iron formulations alone. This can be due to GI intolerance and the inability to maintain iron stores due to blood loss. Therefore, it is not uncommon for patients with HHT to require treatment with intravenous iron formulations. The response to iron can be seen with an increase in the reticulocyte count initially and an improvement in hemoglobin by 4–6 weeks. 34 Once replaced, patients require ongoing monitoring of anemia and iron studies. The time interval between measurements depends on prior experience with a patient and the current status of ongoing bleeding. Failure to maintain iron reserves using intravenous iron indicates the use of additional therapies to reduce bleeding.
Hematologists also assist other clinicians in the management of bleeding in patients with HHT. Although patients can self-report increased epistaxis or melena, hematologists can identify patients who need additional interventions by tracking intravenous iron and blood transfusion needs. Patients are then referred to the appropriate collaborating specialties, such as ear, nose, and throat (ENT) or gastroenterology, for consideration of local therapies.
Patients who do not respond to local therapies or have numerous bleeding sites that are not amenable to local intervention can consider systemic therapies. Standard systemic therapies include antifibrinolytic agents, hormonal therapies, and antiangiogenic agents. 29 Antiangiogenic agents are generally well tolerated and are associated with significant reduction in bleeding and transfusions.28,29 Patients should be encouraged to participate in clinical trials designed to study and optimize current therapies and identify novel therapeutic interventions. Accepted and experimental treatment strategies involve an agreement among all members of the multidisciplinary HHT team.
Pulmonary AVMs and other cardiopulmonary manifestations
One or more PAVM is identified in 20–50% of patients with HHT. 35 They are found most commonly with the HHT1/ENG mutation, but all HHT cases are at risk. Clinical features attributable to complications of PAVM such as stroke, brain abscess, or hemoptysis may be the sentinel manifestation of HHT. Yet most PAVMs are asymptomatic, especially when small, until a complication arises. Thus, screening for PAVMs is recommended in suspected and confirmed HHT to prevent serious sequelae, 7 and is currently the standard of care at North American accredited centers. 36 Transthoracic saline contrast echocardiography (‘bubble echo’) is the preferred initial screening test for pulmonary AVM. 7 In cases of grade 2 to 3 shunting or a high suspicion of PAVM with low grade (grade 1) or indeterminate shunts, we perform contrast-enhanced chest computed tomography angiography (CTA) using a modified pulmonary embolism protocol with a low-dose technique. The use of intravenous contrast with CTA allows a more definitive evaluation of the vascular anatomy, can detect cases of partial AVM thrombosis, and helps evaluate PAVM mimics, including granulomas, nodules, bronchoceles and vascular mimics such as arterial and venovenous collaterals, pulmonary artery pseudoaneurysms, Sheehan vessels, and pulmonary vein varices. As with all intravenous access cases, a wet-to-wet tubing connection is recommended to prevent air embolism.
Once detected, the decision to treat a PAVM depends on the accessibility of the nidus and the diameter of the feeding artery, the patient’s ability to tolerate the procedure, and its inherent risks. Most procedures can be performed with moderate sedation, although anesthesia support is advised in selected cases. In patients with left bundle branch block, external pacing may be used during the procedure. Generally, PAVMs with a feeding artery diameter of > 3 mm are targeted. However, with advances in imaging and catheter technology, there has been a trend to attempt embolization for smaller AVMs, especially if other larger lesions are being treated during the same procedure. 37 Primary contemporary embolic materials are coils and plugs (Figure 6). There is debate on the preferred embolization method; traditionally, the feeding artery was occluded, but some recent investigators advocate sac embolization to improve the occlusion rate. 38 The risk of serious complications from treatment is < 1%, including stroke, catheter access site complications, and pulmonary infarction. Mild pleuritic chest pain is the most common minor complication, reported in up to 13% of patients, and can be managed with nonsteroidal antiinflammatory drugs (NSAIDs). 39 Following successful embolization, CTA is performed to confirm durable AVM occlusion in 6 months, and patients undergo clinical evaluation with our multidisciplinary team. Since previously occluded AVMs can recanalize in up to 20% of patients, or new feeding arteries to occluded AVMs can develop in up to 6% of patients, continued follow-up at 3 to 5-year intervals with CTA is recommended. In patients with poorly accessible lesions or a feeding artery too small for intervention, CTA follow-up can be performed every 5 years, as a tiny proportion will slowly enlarge and smaller lesions tend to remain stable. 40 If the initial screening CTA is negative for PAVM but a shunt is present on contrast echocardiography, microscopic AVMs may be present. These patients can be followed with 3 to 5-year CTA. Given the sensitivity of CTA for the detection of AVMs, pulmonary angiography is typically only performed for therapy.
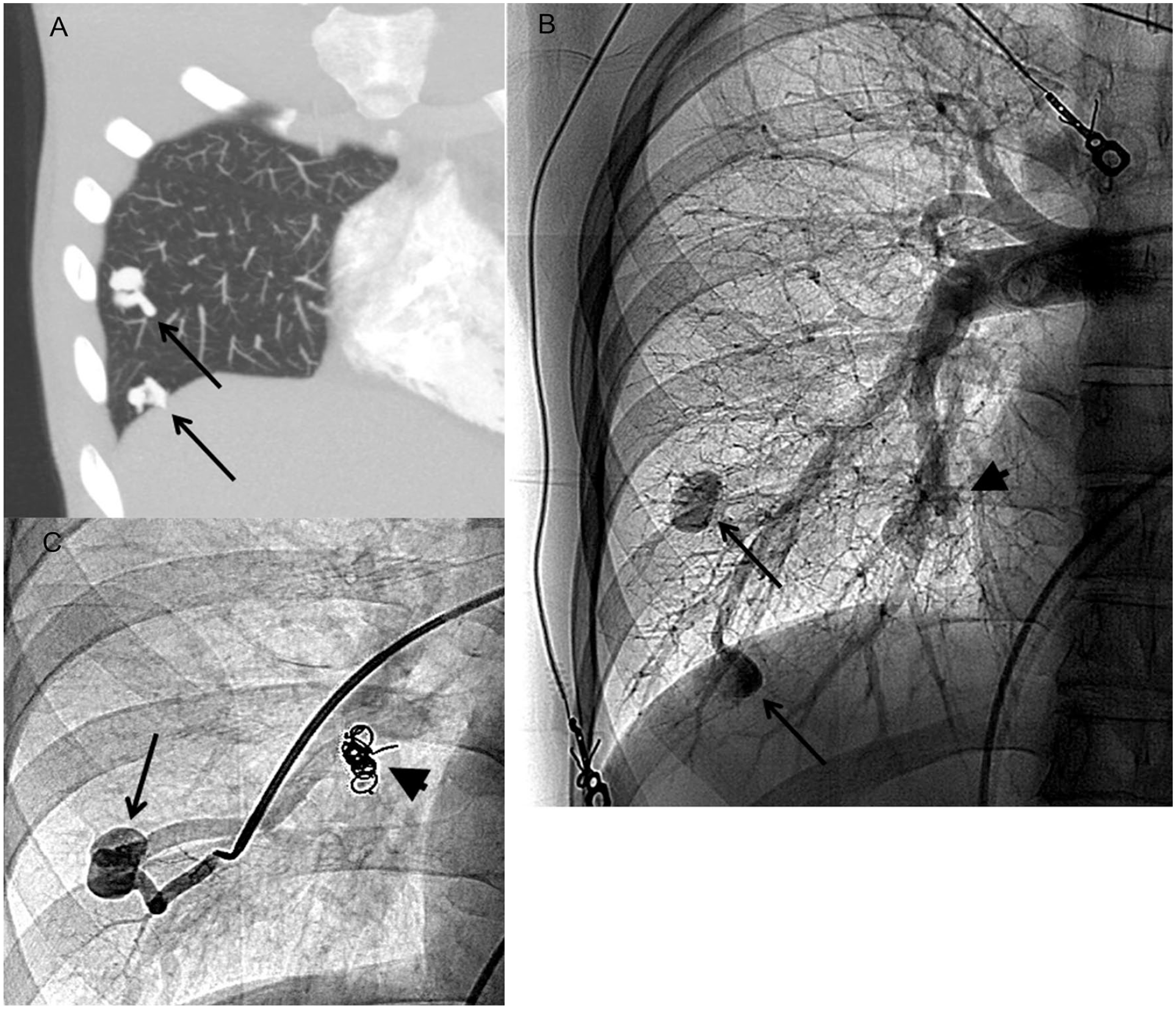
Pulmonary arteriovenous malformation in a patient with HHT.
Beyond the most commonly recognized PAVMs, other pulmonary complications of HHT include pulmonary hypertension resulting from high output heart failure or associated with liver (portal hypertension and/or hepatic dysfunction), and venous thromboembolism.31,41–44 At our center, asthma is frequently seen, likely due to its prevalence in the general population. Patients with pulmonary manifestations of HHT most commonly report dyspnea, with hypoxemia observed in a minority of patients. The evaluation of patients with pulmonary symptoms depends on the clinical setting. Generally, it includes pulse oximetry at rest and with exercise, a 6-minute walk distance, bubble echo study, transesophageal echocardiogram, chest (cardiac and pulmonary) CT angiogram, and a complete set of pulmonary function tests.
For patients with HHT with heart failure, a transthoracic echocardiogram should be performed to assess right and left ventricular size and function, and the presence of pulmonary hypertension and high cardiac output should be confirmed by hemodynamic measurements in the cardiac catheterization laboratory. Treatment of pulmonary hypertension and high cardiac output heart failure includes therapies for closing or reducing left to right shunting from AVMs with or without heart transplantation. 45
In patients with HHT with coronary artery disease with or without intracoronary stents, long-term dual antiplatelet therapy with aspirin and P2Y12 inhibitor is generally contraindicated in high-risk HHT patients due to bleeding complications.46,47 Consequently, percutaneous coronary interventions should be avoided and patients should be managed medically as much as possible. If coronary stents are required, dual antiplatelet therapy is usually given for 1 month followed by continuation of P2Y12 inhibitor for 2 additional months, and then only aspirin if tolerated without worsened epistaxis or GI bleeding.
Venous thromboembolism is not a specific manifestation of HHT. In patients with HHT who develop lower-extremity deep venous thrombosis from other causes, an inferior vena cava filter is recommended with minimal or without anticoagulation, so as not to worsen epistaxis and other HHT-related bleeding. For patients at risk for cardioembolic events from atrial fibrillation or left ventricular thrombus, the risk of subsequent events without anticoagulation should be weighed against the risk of bleeding from HHT with anticoagulation therapy in determining the course of action. Some patients with HHT can tolerate anticoagulation therapy without severe bleeding. 48 Ablation procedures may be considered to minimize or eliminate anticoagulation for atrial fibrillation in cases not tolerating anticoagulation.
Gastrointestinal manifestations
Blood loss in HHT is predominantly due to epistaxis. GI bleeding occurs in 13–30% of patients but rarely before age 50 years. 49 Arteriovenous malformations of HHT can occur throughout the GI tract, most commonly the stomach and proximal small intestine. AVMs of the skin and GI tract can occur later in life; therefore, HHT must be considered in adult patients with bleeding and multiple AVMs in the GI tract. It can be difficult to distinguish blood loss from epistaxis versus GI bleeding. Both can present as iron deficiency anemia or melena. GI bleeding is suspected when blood loss is out of proportion to that occurring from nosebleeds. In children or young adults, juvenile polyps that occur in those with the SMAD4 mutation are a more likely cause of GI bleeding. Adults over 45 years of age with iron deficiency anemia require a colonoscopy to assess for colon cancer.
When GI bleeding is suspected, an upper endoscopy is performed (Figure 7). The best modality for therapy of an actively bleeding AVM in the GI tract has not been determined. Endoscopic argon plasma coagulation thermal therapy is commonly used, but a clip may be a better approach to avoid excessive thermal injury. There is no evidence that repeated thermal ablation of nonbleeding AVMs is of benefit, and it may be harmful. If no bleeding source is found during upper endoscopy, a small bowel capsule endoscopy should be performed to localize the bleeding site.
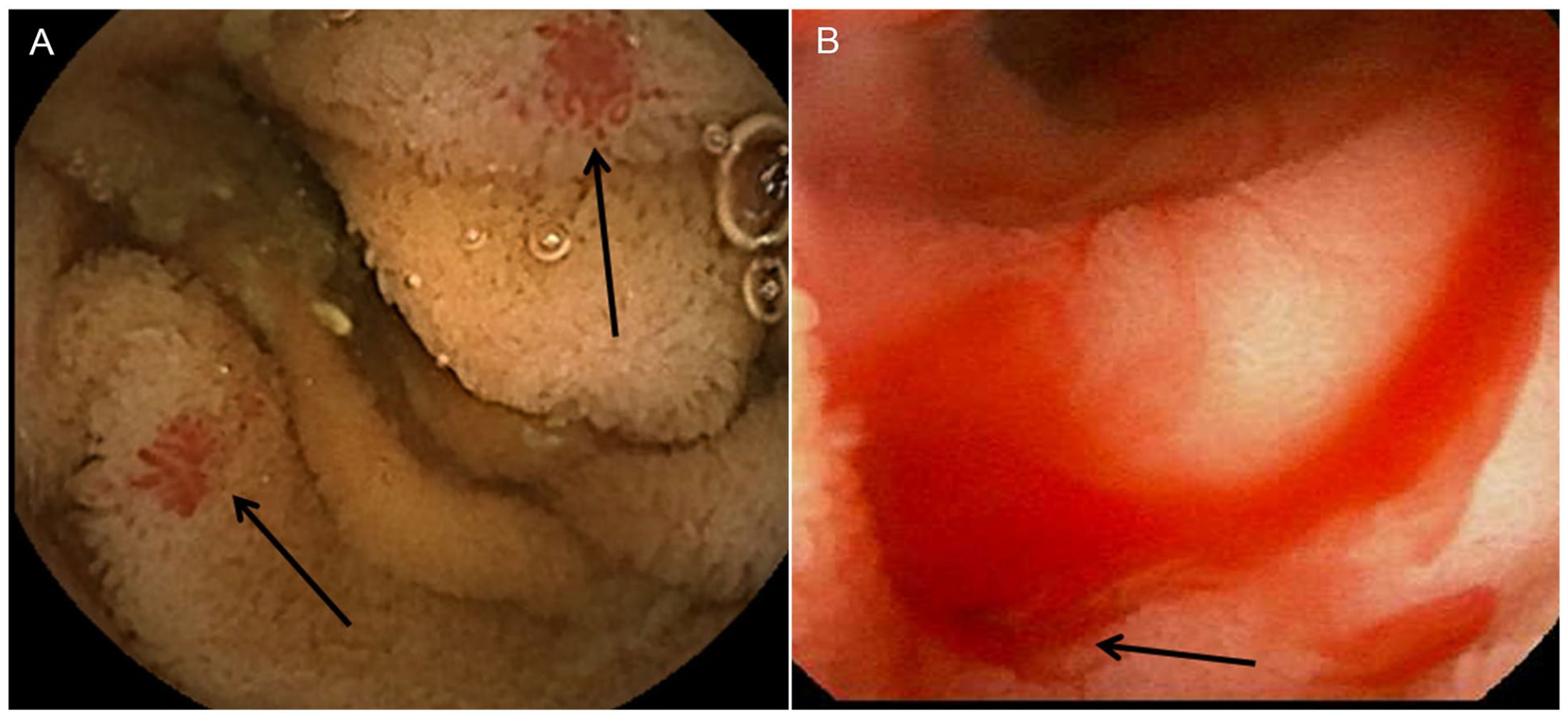
Mucosal AVM of the small intestine in a patient with HHT as seen by upper GI endoscopy.
Therapy for suspected GI bleeding is based on the magnitude of the blood loss and is defined as (1) mild bleeding and hemoglobin maintained on oral iron; (2) moderate bleeding and hemoglobin maintained with IV iron; and (3) severe bleeding and unable to maintain hemoglobin with iron therapy and/or needs transfusions.7,49,50
Treatment for mild suspected GI bleeding is oral antifibrinolytic agents. The use of systemic antiangiogenic therapy is considered for moderate to severe GI bleeding in conjunction with endoscopic therapy, and this is presented in the hematology section of this review.
Juvenile polyposis syndrome
A clinical diagnosis of juvenile polyposis syndrome (JPS) is made when any one of the following is met: five or more juvenile polyps in the colon, multiple juvenile polyps in the GI tract, or any number of juvenile polyps with a family history of JPS. Juvenile polyps are classified as hamartomas but could predispose to colon cancer. About 50% of individuals who meet clinical criteria have pathogenic variants in one of the two genes involved in TGFß signaling, SMAD4, and BMPR1A. The primary clinical manifestations are juvenile polyps and cancers in the colon and stomach. JPS cases who are SMAD4 carriers are also at risk for other manifestations of HHT, including epistaxis, PAVMs, and BAVMs. In a multicenter study of JPS, SMAD4 carriers (compared to carriers with BMPR1A) had a higher prevalence of anemia (58% vs 26%), other HHT manifestations (32% vs 0%), and gastric polyps (39% vs 13%). 51 Colonic polyps were found in both BMPR1A and SMAD4 index carriers (91% and 86%) with proximal distribution and numbering between five and 100. The overall cancer rate was 15%, with 78% identified before or at the time of JPS diagnosis. Colorectal cancer was found in 12% and 7% of carriers of SMAD4 and BMPR1A, respectively.
Current guidelines recommend management in a specialized center with colonoscopy and upper endoscopy every 2–3 years (or more frequently based on polyps or symptoms) starting at age 12–15 years. 52 Carriers of SMAD4 pathogenic mutations should also be screened for HHT multisystem involvement beginning within the first 6 months of life or at the time of their genetic diagnosis.
Liver AVMs
Liver AVMs affect about half of HHT cases but rarely cause serious sequelae nor require intervention. 53 Abdominal imaging is not routinely recommended, 7 unless liver function tests are abnormal on screening laboratory tests. Liver AVMs in patients with HHT can range from small, clinically insignificant, to those leading to liver failure, high output cardiac failure, or severe pulmonary hypertension. 54 The likelihood of needing a liver transplant in patients with liver AVMs is rare. This would be considered in patients with symptomatic complications, including cardiac failure, clinically significant portal hypertension, biliary complications, or liver failure.
Neurologic manifestations
Neurological manifestations occur in 8–10% of patients with HHT. The etiologies of these manifestations can be divided into two categories: those secondary to PAVMs and those due to CNS vascular malformations. 55
The most frequent complications of PAVMs include ischemic strokes and cerebral abscesses. Strokes affect roughly 30% of patients with HHT, 55 often involving multiple arterial territories, which can be appreciated on MRI. These are attributed to paradoxical embolism, with clots arising in the systemic venous circulation passing unfiltered through the lung to the cerebral arterial circulation, through shunting in PAVMs. Cerebral abscesses are thought to also arise from a similar mechanism of paradoxical emboli arising with transient bacteremia. Brain abscesses were noted in 5–10% of patients in early series of HHT before routine screening and aggressive management of PAVMs. Prevention of these complications is focused on the early detection and treatment of PAVMs. 56 Once PAVMs are treated, the occurrence of strokes and cerebral abscesses in HHT is rare. With current screening and aggressive treatment of PAVMs, we have not noted any cases of septic brain abscess in our HHT center cohort. Few cases who presented with ischemic stroke or a history of such stroke from PAVM in our center had not undergone prior HHT evaluation.
BAVMs account for one-third of neurological manifestations in patients with HHT. 57 These are commonly multiple in the setting of HHT, and AVM multiplicity in the brain should raise a diagnostic consideration of HHT. The annual risk of BAVM rupture is estimated to be less than 1% per year, and is greater in symptomatic lesions, particularly those that caused prior hemorrhage, and in lesions with progression of angiographic features (growth of nidus or acquisition of new aneurysms). The rupture of a BAVM results in a hemorrhagic stroke, which may present as headache, seizures, focal neurological deficit, or devastating brain injury, depending on the size and location of the lesion.
MRI screening is considered in adult patients with possible or definite HHT to detect cerebral AVMs, and this is currently recommended at all accredited HHT centers of excellence in the United States. 7 At our center, we advocate MRI of the brain with and without contrast to screen for cerebral AVMs, including susceptibility weighted imaging to maximize sensitivity and time-of-flight MRA. Children with headaches or other neurologic symptoms should be similarly screened. Screening for BAVMs in asymptomatic children may otherwise be deferred until they may undergo MRI without anesthesia. At our institution, patients without a vascular malformation on brain MRI/MRA are recommended for repeat screening every 10 years to detect any potential de novo development of AVMs.
The management of BAVMs depends on the location and size of the lesion, symptoms, and life expectancy. Treatment options include close observation for any progression of the AVM (typically with annual MRI/MRA surveillance), or proactive treatment. Treatment is indicated in cases of AVM bleeding, and is considered in younger patients, those with associated symptoms, and larger lesions associated with aneurysm or varix. Treatment may involve therapeutic embolization, stereotactic radiation, and/or microsurgical resection, with the goal of complete obliteration of the AVM (Figure 8). The HHT guidelines recommend an individualized approach by a multidisciplinary team with neurovascular expertise. 7
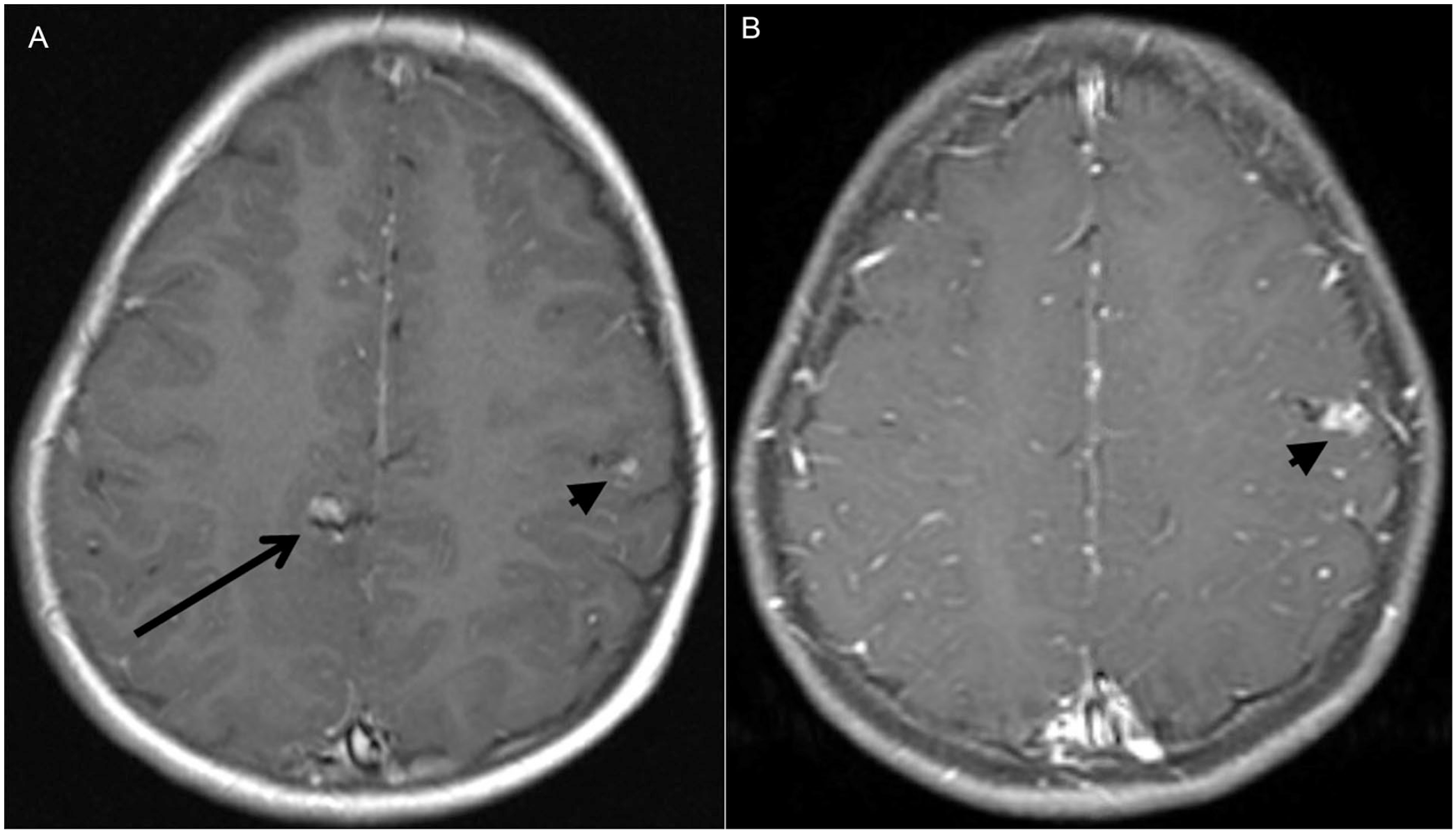
Multiple brain AVMs in a patient with HHT.
Patients with HHT may present with seizures related to prior neurologic sequelae, or with headache syndrome. These are managed symptomatically with anticonvulsant medications or headache regimen, after excluding the presence of an untreated AVM, particularly one that has changed over time or could explain the symptoms.
Skin, lip, and oral lesions
Skin and oral small punctate vascular lesions commonly referred to as telangiectasias in this syndrome, are typical features of HHT, with a published incidence ranging between 50% and 80%. However, if sites beyond those identified in the Curaçao criteria (lips, fingers, oral cavity) are included, the incidence may be much higher.58,59 Skin and oral lesions increase with age but can be detected as early as infancy with careful examination. 60 On exam, the telangiectasias are generally small punctate bright red macules and papules (Figure 1). Histologically, these lesions are tiny AVMs. 17 Symptomatic bleeding is very rarely associated with these punctate lesions. Vascular lesions of cosmetic concern are brought to the attention of dermatologists or oral surgeons for discussion of ablative interventions.
Pulsed dye laser ablation is a generally effective approach to treatment. 61 This minimally invasive outpatient procedure can be performed for both cutaneous and lip lesions. Often, multiple treatments are needed for resolution. The most common adverse event is postprocedure bruising, but in general, the procedure requires little recovery time for the individual. Needle cautery is another modality that may be used for ablation of telangiectasias, though it has a higher risk of scarring than vascular laser therapy.
Laser or electrocautery treatments can be perceived as painful and anxiety-provoking in the pediatric population; therefore, it is advised to delay such procedures until the child is personally invested and motivated to undergo treatment. Additional skin-directed management focuses on sun protection to minimize dilation and accentuation of the telangiectasias.
Conclusion
Patients with HHT mostly dread being evaluated by physicians who do not know the disease and have limited experience with its management. Other concerns are uncoordinated care, where one specialist may be unaware of HHT problems in other systems, or where the patient is not properly screened or treated for a serious HHT-related problem. And finally, many patients face problems with insurance coverage for proper HHT care. Accredited HHT centers aim to mitigate these problems. And they allow exchange of information among centers on less common disease manifestations, and the implementation of novel or experimental protocols.
Though evidence-based management guidelines have been published, this review provides practical recommendations on the implementation of practical multidisciplinary protocols for disease management (Table 2).
Key points to remember.

AVMs, arteriovenous malformations; HHT, hereditary hemorrhagic telangiectasia; JPS, juvenile polyposis syndrome.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Data availability
The HHT Center of Excellence at University of Chicago intake forms for patient screening and electronic medical record tools for cross notification are available upon request.