
Abstract
Background:
This study evaluated the association of smoking with mitochondrial function in gastrocnemius muscle of people with peripheral artery disease (PAD).
Methods:
Participants were enrolled from Chicago, Illinois and consented to gastrocnemius biopsy. Mitochondrial oxidative capacity was measured in muscle with respirometry. Abundance of voltage-dependent anion channel (VDAC) (mitochondrial membrane abundance), peroxisome proliferator-activated receptor-γ coactivator (PGC-1α) (mitochondrial biogenesis), and electron transport chain complexes I–V were measured with Western blot.
Results:
Fourteen of 31 people with PAD (age 72.1 years, ABI 0.64) smoked cigarettes currently. Overall, there were no significant differences in mitochondrial oxidative capacity between PAD participants who currently smoked and those not currently smoking (complex I+II-mediated oxidative phosphorylation: 86.6 vs 78.3 pmolO2/s/mg, respectively [p = 0.39]). Among participants with PAD, those who currently smoked had a higher abundance of PGC-1α (p < 0.01), VDAC (p = 0.022), complex I (p = 0.021), and complex III (p = 0.021) proteins compared to those not currently smoking. People with PAD who currently smoked had lower oxidative capacity per VDAC unit (complex I+II-mediated oxidative phosphorylation [137.4 vs 231.8 arbitrary units, p = 0.030]) compared to people with PAD not currently smoking. Among people without PAD, there were no significant differences in any mitochondrial measures between currently smoking (n = 5) and those not currently smoking (n = 63).
Conclusions:
Among people with PAD, cigarette smoking may stimulate mitochondrial biogenesis to compensate for reduced oxidative capacity per unit of mitochondrial membrane, resulting in no difference in overall mitochondrial oxidative capacity according to current smoking status among people with PAD. However, these results were cross-sectional and a longitudinal study is needed.
Introduction
Compared to people without peripheral artery disease (PAD), those with PAD have pathophysiologic changes in gastrocnemius muscle including reduced muscle mass, increased fatty infiltration, and abnormal mitochondrial structure.1,2 Skeletal muscle abnormalities are associated with walking impairment and mobility loss in PAD. 2 Sustained skeletal muscle function requires energy produced by oxidative phosphorylation, which is carried out by the electron transport chain (ETC), a protein complex in the inner membrane of mitochondria. 3
Preliminary evidence suggests that cigarette smoke impairs mitochondrial respiratory complex activity in skeletal muscle.3–9 Despite high rates of smoking in people with PAD, 10 to our knowledge, associations of smoking and mitochondria health have not been assessed in PAD. This study evaluated the association of cigarette smoking with mitochondrial respiratory function and mitochondrial protein abundance in gastrocnemius muscle biopsies of people with PAD.
Methods
Study design
Participants were part of an observational study of mitochondrial activity, measured with gastrocnemius muscle biopsy, and functional performance in people with and without PAD. Data were collected between July 23, 2018 and August 9, 2021. The Institutional Review Board of Northwestern University approved the protocol. All participants provided written informed consent.
Identification of participants
Potential participants were identified from lists of patients with and without PAD obtained from Northwestern Memorial Hospital, advertisements on Chicago Transit Authority buses and trains, and mailed postcards to people 55 and older living in the Chicago area. People who participated in prior studies with a study investigator (MMM) and expressed interest in future research were contacted.
Inclusion and exclusion criteria
PAD was defined as an ankle–brachial index (ABI) ⩽ 0.90 in either leg. 11 Absence of PAD was identified as an ABI of 1.00–1.40. To balance the mean age between those with and without PAD, only people without PAD aged 60 and older were eligible.
Potential participants were excluded if they had an ABI of 0.90–0.99 indicating mild lower-extremity atherosclerosis,11–13 a Mini-Mental State Examination score < 23, 14 a major medical illness, or planned major surgery; or if they were participating in a clinical trial or unwilling to undergo a gastrocnemius biopsy. Potential participants with PAD were excluded if they had chronic limb-threatening ischemia or walking limitations by a condition other than PAD. Potential participants without PAD who had diabetes were excluded.
ABI measurement
Systolic pressures were obtained twice in each brachial, dorsalis pedis, and posterior tibial artery using a hand-held Doppler probe (Pocket Dop II; Nicolet Biomedical, Inc., Golden, CO, USA). The ABI was calculated by dividing the mean of the dorsalis pedis and posterior tibial pressures in each leg by the mean of the four brachial pressures. 15
Gastrocnemius muscle biopsy
Open muscle biopsy was performed in the medial head of the gastrocnemius muscle of the leg with the lower ABI.16,17 Anesthesia was achieved with subcutaneous lidocaine. Subcutaneous and adipose tissue were dissected. Approximately 250 mg of muscle tissue were removed, of which 10–20 mg were stored in cold biopsy media for respirometry, and the remainder immediately prepared for freezing at −80°C.
Mitochondrial oxidative capacity measurement
High-resolution respirometry was performed with an Oroboros O2k (Oroboros Instruments, Innsbruck, Austria) using established protocols.18,19 Muscle biopsies were separated in ice-cold preservation media (BIOPS) under a dissecting microscope to obtain four 2–3 mg replicates per participant, which were permeabilized with saponin for 20 minutes, then washed in mitochondrial respiration media (MiR05) for 10 minutes. All data were collected at 37°C under hyperoxygenated (200–450 µM O2) conditions in MiR05 to avoid limitations with oxygen diffusion. The carbohydrate and fatty acid substrate-uncoupler-inhibitor titration (SUIT) protocols used to test for respiration were: malate, pyruvate or octanoylcarnitine, ADP, glutamate, cytochrome c, succinate, followed by titrations of carbonyl cyanide m-chlorophenyl hydrazine (CCCP), rotenone, and antimycin A. Results from the four replicates that differed by more than 15% following the addition of cytochrome c were considered over-permeabilized and were not included in analyses. The state of respiration after adding glutamate was defined as complex I(+III+IV+ATP synthase)-mediated maximal oxidative phosphorylation capacity (PCI). Respiration after succinate addition was defined as complex I+II(+III+IV+ATP synthase)-mediated (PCI+II), effectively maximal, oxidative phosphorylation capacity. The state of respiration after adding CCCP (uncoupler) was considered maximal ETC capacity (ECI+II). Fatty acid respiration was measured using complex I(+III+IV+ATP synthase)-mediated oxidative phosphorylation (PCI/FA) by adding malate, octanoylcarnitine, and ADP substrates.
Western blot for mitochondria-associated proteins
Protein expression of mitochondrial ETC complexes I–V; voltage-dependent anion channel (VDAC), an abundant outer membrane protein that facilitates passage of metabolites into and out of the mitochondrion and is used to approximate mitochondrial quantity; and peroxisome proliferator-activated receptor-γ coactivator (PGC-1α), a protein that stimulates mitochondrial biogenesis; were measured by Western blot19,20 using Total oxidative phosphorylation Human WB Antibody Cocktail (1:2000; Abcam, ab110411; Waltham, MA, USA) for subunits of complexes I–V, anti-VDAC (1:1000; Cell Signaling, #4661s; Danvers, MA, USA), and anti-PGC-1α (1:1000; Millipore/Sigma, #516557; Burlington, MA, USA) as primary antibodies (Figure 1). Densitometry of the target bands on the immunoblots was performed and quantified using Image Lab™ (version 6.0.1; Bio-Rad Laboratories, Hercules, CA, USA). The target bands were normalized to the total protein content of the sample as measured by Ponceau-S staining of the immunoblot. Several gels were needed to accommodate the number of samples, and on each the same cross-sample was run in parallel to participant samples. The protein-load-normalized target band densities were normalized to the cross-sample’s target band on the same immunoblot to account for technical inter-membrane differences.
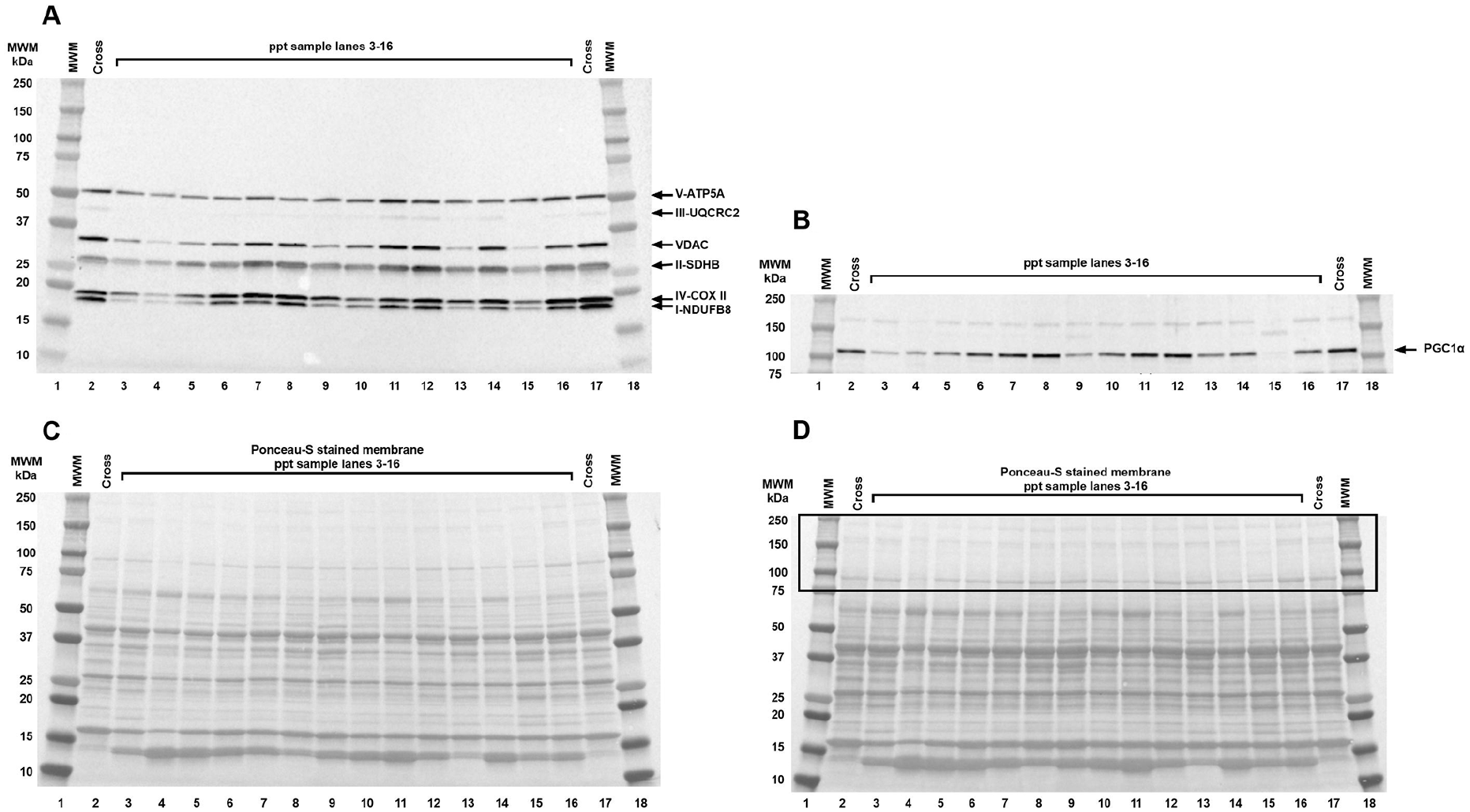
Representative immunoblot of protein subunits of the mitochondrial electron transport chain complexes, VDAC (A and C) and PGC-1α (B and D), in a muscle biopsy sample from a study participant.
Additional measures
Following a standardized protocol, participants walked along a 100-foot hallway for 6 minutes, covering as much distance as possible.21,22 The distance completed after 6 minutes was recorded. Free-living physical activity was measured over 7 days with the ActiGraph accelerometer and recorded as total counts per day. The accelerometer was worn on the right hip and removed only for bathing or sleeping.23,24
Height and weight were measured at the study visit. Body mass index (BMI) was calculated as weight (kg)/height (m2). Comorbidities were self-reported based on whether a physician had previously diagnosed the participant with each comorbid condition. 25 Race was self-reported in response to an open-ended question. Current cigarette smoking status and pack-year history were self-reported as part of a health history questionnaire. Former smokers were defined as those who selected: ‘Yes, but have now quit smoking’.
Statistical analysis
Baseline characteristics were summarized as means and SDs for continuous variables and as frequencies and percentages for categorical variables. Two-sample t-tests were used to compare continuous variables, and chi-squared or Fisher’s exact tests were used to compare categorical variables between participants who currently smoked and those who did not, separately among participants with and without PAD.
Muscle measurements were summarized as means and SDs for continuous variables. Two-sample t-tests were used to compare continuous variables between participants who currently smoked with those who did not, separately among participants with and without PAD. The comparison of participants with PAD by current smoking status was repeated via analysis of covariance adjusting for age, sex, race, ABI, and BMI, which were selected due to either their potential associations with muscle measurements or observed imbalances between people who currently smoked and people who did not currently smoke. These variables were selected for adjustment prior to conducting the analysis of covariance.
Analysis of covariance was used to compare muscle measurements between current, former, and never smokers among participants with PAD. A trend test was performed via a linear regression analysis with muscle measurements as dependent outcomes and smoking groups (never smokers, former smokers, and current smokers) as ordinal independent variables.
The goodness-of-fit test for normality distribution (Kolmogorov–Smirnov test) was conducted for each outcome by groups. If there was statistically significant evidence against the normal assumption, the nonparametric Mann–Whitney rank test was used for two-group comparisons, proportional odds regression for ordinal outcomes was used for adjusting for baseline covariates, and the Kruskal–Wallis test was used for analysis for variance.
All statistical tests were two-sided at the statistical significance level of 0.05. Statistical analyses were conducted using SAS 9.4 (SAS Institute Inc., Cary, NC, USA); RStudio 2022.07.2+576 (RStudio, PBC, Boston, MA, USA) was used for proportional odds regression.
Results
Participants with PAD
Among 31 people with PAD (mean 72.1 years, mean ABI: 0.64), 14 reported currently smoking cigarettes (Table 1). Participants with PAD who currently smoked were younger (p = 0.048) and had a lower prevalence of angina (p = 0.048) compared to participants with PAD who did not currently smoke (Table 1). There were no differences in functional performance measures between participants with PAD who currently smoked and participants with PAD who did not currently smoke (online Supplemental Table 1).
Characteristics of people with and without PAD by current cigarette smoking status.
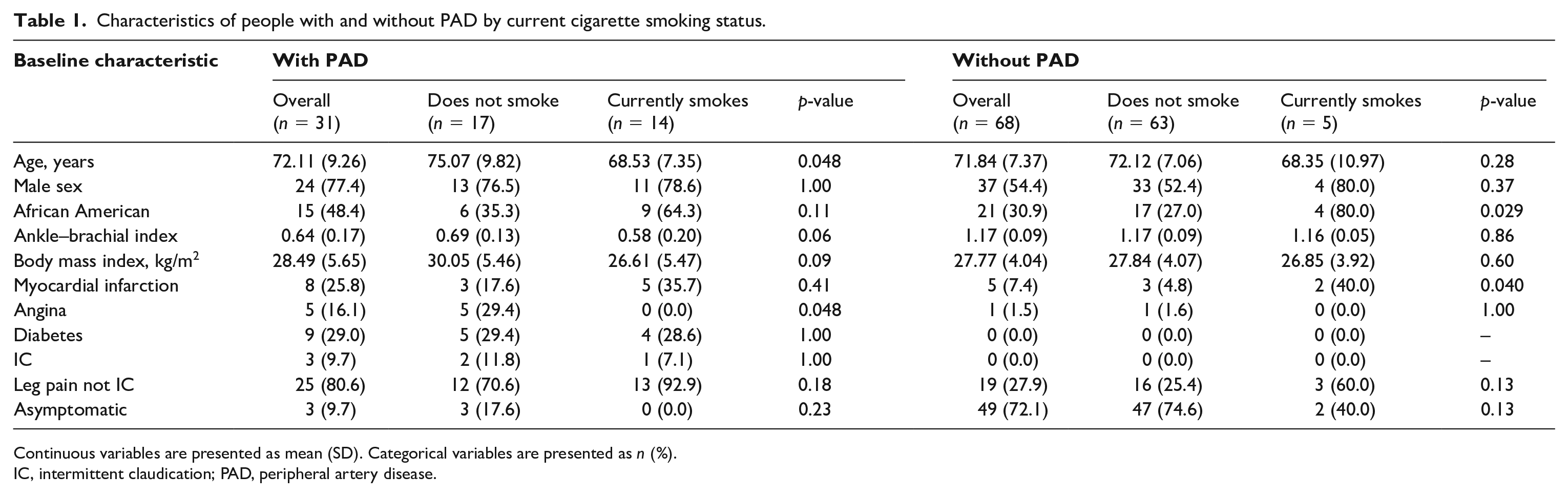
Continuous variables are presented as mean (SD). Categorical variables are presented as n (%).
IC, intermittent claudication; PAD, peripheral artery disease.
Participants with PAD who currently smoked had a greater abundance of PGC-1α (p = 0.009), VDAC (p = 0.022), complex I (p = 0.021), and complex III (p = 0.021) in gastrocnemius specimens compared to participants with PAD who did not currently smoke (Table 2). After statistical adjustment for age, sex, race, ABI, and BMI, associations were attenuated but participants who currently smoked had a statistically significant higher abundance of VDAC (p = 0.037) and complex I (p = 0.017) compared to participants with PAD who did not currently smoke (Table 2).
Gastrocnemius mitochondrial measures by current cigarette smoking status among people with peripheral artery disease.
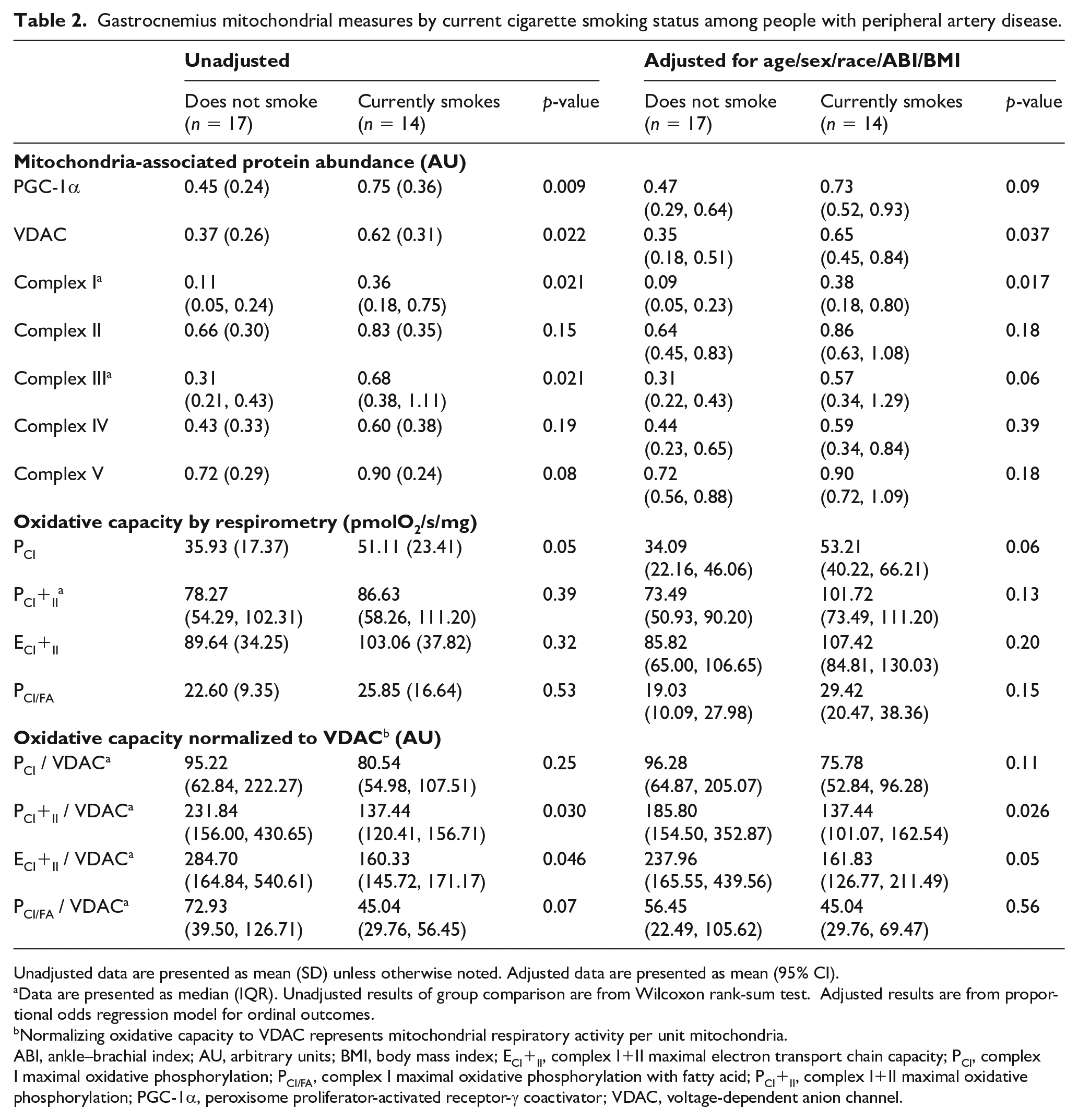
Unadjusted data are presented as mean (SD) unless otherwise noted. Adjusted data are presented as mean (95% CI).
Data are presented as median (IQR). Unadjusted results of group comparison are from Wilcoxon rank-sum test. Adjusted results are from proportional odds regression model for ordinal outcomes.
Normalizing oxidative capacity to VDAC represents mitochondrial respiratory activity per unit mitochondria.
ABI, ankle–brachial index; AU, arbitrary units; BMI, body mass index; ECI+II, complex I+II maximal electron transport chain capacity; PCI, complex I maximal oxidative phosphorylation; PCI/FA, complex I maximal oxidative phosphorylation with fatty acid; PCI+II, complex I+II maximal oxidative phosphorylation; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator; VDAC, voltage-dependent anion channel.
In unadjusted analyses and analyses adjusted for age, sex, race, ABI, and BMI, there were no significant differences in mitochondrial oxidative capacity among participants with PAD by current smoking status (Table 2). However, after adjusting for mitochondrial membrane abundance by normalizing to VDAC, participants with PAD who currently smoked had significantly lower PCI+II (p = 0.030) and ECI+II (p = 0.046) compared to participants with PAD who did not currently smoke (Table 2). The association in ECI+II normalized to VDAC was attenuated and no longer significant after statistical adjustment for age, sex, race, ABI, and BMI (Table 2).
Across current, former, and never smokers with PAD there were statistically significant linear trends in complex I abundance (p = 0.032) and complex III abundance (p = 0.026), with current smokers having a higher abundance of these protein complexes compared to never smokers. Similar findings were observed for PGC-1α abundance (p = 0.026) (online Supplemental Table 2). There were no statistically significant differences in mitochondrial oxidative capacity between current, former, and never smokers with PAD (online Supplemental Table 2).
Participants without PAD
Among 68 participants without PAD (mean 71.8 years, mean ABI: 1.17), five reported currently smoking cigarettes (Table 1). There were no statistically significant differences in mitochondrial protein abundance nor oxidative capacity, before or after normalization to VDAC, among people without PAD by current smoking status, though sample size was small (Table 3).
Gastrocnemius mitochondrial measures by current cigarette smoking status among people without peripheral artery disease.
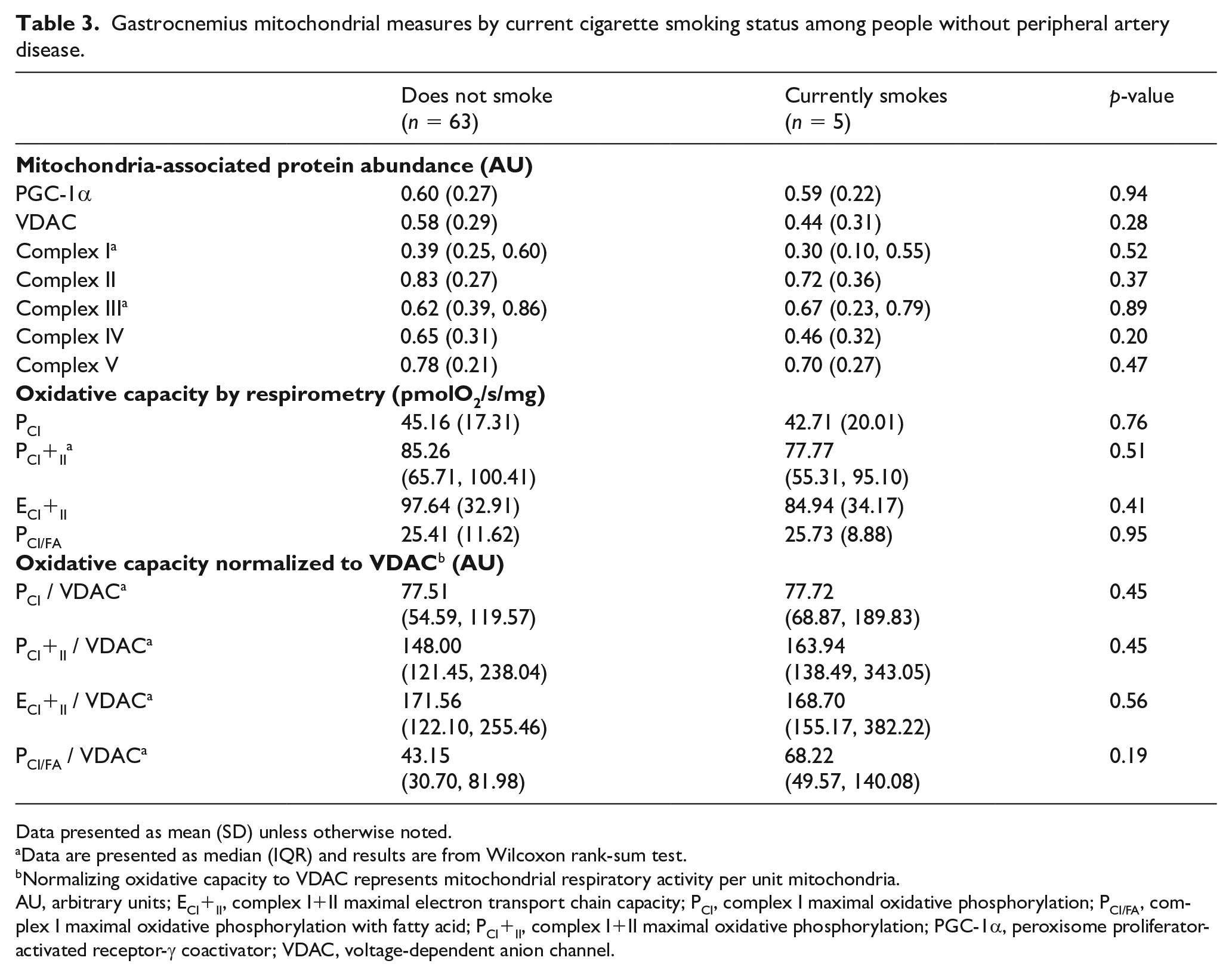
Data presented as mean (SD) unless otherwise noted.
Data are presented as median (IQR) and results are from Wilcoxon rank-sum test.
Normalizing oxidative capacity to VDAC represents mitochondrial respiratory activity per unit mitochondria.
AU, arbitrary units; ECI+II, complex I+II maximal electron transport chain capacity; PCI, complex I maximal oxidative phosphorylation; PCI/FA, complex I maximal oxidative phosphorylation with fatty acid; PCI+II, complex I+II maximal oxidative phosphorylation; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator; VDAC, voltage-dependent anion channel.
Discussion
In this study, among people with PAD, people who currently smoked cigarettes had a significantly greater abundance of PGC-1α, a protein marker associated with mitochondrial biogenesis, compared to those who did not smoke. Among people with PAD, those who currently smoked cigarettes had greater abundance of the outer mitochondrial membrane protein VDAC, a marker of increased mitochondrial content overall, and greater abundance of mitochondrial complexes I and III, compared to people who did not currently smoke. Though these findings would typically indicate greater oxidative capacity in participants with PAD who currently smoked cigarettes given the role of ETC complexes in energy production, there was no overall difference in oxidative capacity between people with PAD who smoked cigarettes and people with PAD who did not smoke cigarettes. Instead, mitochondrial oxidative capacity per unit of mitochondrial membrane was significantly lower in participants with PAD who currently smoked compared to participants with PAD who did not currently smoke, indicating a reduction in mitochondrial quality and amount of energy produced per mitochondrion among people with PAD who smoke cigarettes. Together, results suggest that people with PAD who currently smoke have increased mitochondrial biogenesis resulting in greater mitochondrial abundance, which appeared to compensate for less efficient mitochondrial oxidative phosphorylation, compared to people with PAD who did not currently smoke.
Prior studies suggested that reduced tissue perfusion and greater hypoxia may explain poorer mitochondrial function in animal models exposed to cigarette smoke and in people without PAD who smoke.3,26 In data reported here, respirometry was performed with adequate oxygen and fuel substrates, yet mitochondrial oxidative capacity, adjusted for mitochondrial membrane abundance, was significantly lower in people with PAD who currently smoked compared to people with PAD who did not currently smoke. These results support the view that, beyond perfusion limitation and hypoxia, there may be intrinsic damage to mitochondria—possibly from long-standing ischemia—causing mitochondrial dysfunction in people with PAD who smoke. Other studies of chronic obstructive pulmonary disease animal models 27 and sedentary humans without PAD 4 suggested that physical deconditioning in people who smoke may explain the association of smoking with mitochondrial oxidative derangements. However, results reported here showed no significant difference in physical activity levels by current smoking status among people with PAD.
There are at least two potential clinical consequences of the findings reported here. First, compensatory increases in abundance of mitochondria in gastrocnemius muscle of current smokers with PAD could overcome deficits in mitochondria quality and facilitate maintenance of skeletal muscle functioning and walking performance. Consistent with this hypothesis, participants with PAD who smoked cigarettes did not have greater functional impairment than participants with PAD who did not smoke cigarettes, even though the skeletal muscle pathology described here suggests greater skeletal muscle mitochondrial impairment in people with PAD who smoke compared to those who do not smoke. If the upregulation of mitochondrial proteins facilitates maintenance of walking performance and delays onset of ischemic symptoms in people with PAD who smoke, this might explain the fact that patients who quit smoking remain at higher risk for a later diagnosis of PAD, particularly if the upregulation of mitochondrial proteins mitigates symptoms of ischemia in people with PAD who smoke. Second, since cigarette smoking is a major risk factor for PAD, further study is needed to determine whether smoking-related muscle damage might contribute to the pathophysiology of muscle abnormalities observed in people with PAD who smoke, independently of lower-extremity ischemia.
Study limitations
This study had several limitations. First, the study was cross-sectional. No causal inferences can be made. Second, the sample size was relatively small. Third, though mitochondrial measures in participants without PAD were included for completeness, the small number of participants without PAD who currently smoked prevents definitive conclusions regarding their smoking-associated mitochondrial changes. Fourth, residual or unmeasured confounding may have explained some findings reported here. Fifth, although muscle biopsy specimens are prepared for respirometry measures under light microscopy to ensure a majority composition of skeletal muscle fibers, other residual cell types may have contributed to some of the oxidative phosphorylation results in the respirometry measurements. Sixth, results may not be generalizable to patients with PAD who have limb-threatening ischemia. Though the proportion of patients with classical symptoms of intermittent claudication was relatively small, based on use of the claudication questionnaire, the prevalence of claudication and atypical exertional leg symptoms were comparable to that of other studies of people with PAD identified from either community or medical center settings. 28
Conclusions
These results support the hypothesis that among people with PAD, cigarette smoking may stimulate mitochondrial biogenesis to compensate for reduced oxidative capacity per unit of mitochondrial membrane, resulting in no overall difference in mitochondrial oxidative capacity by current smoking status. Further study is needed to better understand the biology and clinical significance of these findings.
Supplemental Material
sj-pdf-1-vmj-10.1177_1358863X221143152 – Supplemental material for Cigarette smoking and mitochondrial dysfunction in peripheral artery disease
Supplemental material, sj-pdf-1-vmj-10.1177_1358863X221143152 for Cigarette smoking and mitochondrial dysfunction in peripheral artery disease by Michelle Guo, Mary M McDermott, Sudarshan Dayanidhi, Christiaan Leeuwenburgh, Stephanie Wohlgemuth, Luigi Ferrucci, Charlotte A Peterson, Kate Kosmac, Lu Tian, Lihui Zhao, Robert Sufit, Karen Ho, Michael Criqui, Shujun Xu, Dongxue Zhang and Philip Greenland in Vascular Medicine
Footnotes
Data availability statement
Study data are available from Dr Greenland upon reasonable request.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Dr McDermott reports unrelated research support from Helixmith, ArtAssist, Chromadex, ReserveAge, Mars, and Regeneron.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: American Heart Association grant 18SFRN33900142.
Supplementary material
The supplementary material is available online with the article
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.