
Abstract
Resuscitation from hemorrhagic shock induces endothelial dysfunction and activates inflammatory cascades leading to organ damage. Following restoration of blood flow to ischemic vascular beds, leukocyte–endothelium interactions leading to leukocyte infiltration into the vascular wall occur very early due, in part, to the loss of endothelium-derived nitric oxide (NO). The mechanism by which ischemia–reperfusion injury impairs endothelium-derived NO is not completely understood. We hypothesized that inhibition of Rho-kinase could exert beneficial effects following hemorrhagic shock by preserving endothelial function and attenuating leukocyte trafficking in the microcirculation. Using intravital microscopy, we found that resuscitation from hemorrhage acutely increased the number of rolling and adherent leukocytes in the mouse splanchnic microcirculation. Treatment of mice with the Rho-kinase inhibitor fasudil, markedly attenuated leukocyte–endothelium interaction in response to hemorrhage/reinfusion. The beneficial effect of fasudil was not observed in endothelial nitric oxide synthase (eNOS)−/− mice. In conclusion, inhibition of Rho-kinase prevents inflammatory leukocyte trafficking in the microcirculation via an eNOS-dependent mechanism. Our data support a role for Rho-kinase inhibitors in the treatment of ischemia–reperfusion injury.
Introduction
Hemorrhage/reinfusion is a whole body ischemia–reperfusion injury, which disrupts the hemostasis of the cardiovascular system. Among the many vascular beds affected by this acute cardiovascular syndrome, the splanchnic circulation plays a key pathogenic role because it is able to trigger remote organ damage during hemorrhagic shock. 1 Indeed, hemorrhage/reperfusion initiates a robust inflammatory cascade characterized by the upregulation of cytokine expression 2 and infiltration of activated leukocytes into organ tissues. 3 The accumulation of leukocytes in ischemic tissues leads to end organ damage and ultimately organ failure during hemorrhagic shock.4,5
The vascular endothelium of the microcirculation plays a key role in the recruitment of circulating neutrophils during ischemia–reperfusion 6 and hemorrhage/reinfusion. 7 Accordingly, reperfusion following hemorrhage has been associated with endothelial dysfunction characterized by loss of endothelium-derived nitric oxide (NO).3,8–10 Loss of NO and production of cytokines11,12 lead to rapid upregulation of endothelial cell adhesion molecules.7,13 This ultimately increases leukocyte–endothelium interactions7,14 and promotes infiltration of leukocytes into ischemic tissues.3,12 Despite the well-known, beneficial role of endothelium-derived NO in the preservation of vascular function,15,16 the mechanism by which ischemia–reperfusion impairs endothelial nitric oxide synthase (eNOS) activity remains poorly understood.
One of the upstream regulators of eNOS expression and activity is Rho-kinase. Rho-kinase has been shown to play an important role in leukocyte migration during vascular injury. 17 In addition, Rho-kinase negatively regulates eNOS expression by decreasing eNOS mRNA stability. 18 Further studies have shown that inhibition of Rho-kinase activity preserves eNOS expression, 19 enhances acetylcholine-induced eNOS-dependent vessel relaxation during hypoxia in vitro, 20 and reduces cerebral infarct size following cerebral ischemia in vivo.21,22 Therefore, we hypothesized that changes in eNOS may mediate some of the downstream effects of Rho-kinase on leukocyte recruitment during ischemia–reperfusion injury.
Materials and methods
This study was performed in accordance with the NIH guidelines for the use of experimental animals. The Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University has approved all animal protocols.
Hemorrhage/reinfusion model of vascular injury
Wild-type (C57BL/6) mice and eNOS−/− (C57BL/6J-Nos3tm1Unc) mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Male mice (8–14 weeks old and 20–30 g in body weight) were anesthetized with intraperitoneal injection (ip) of sodium pentobarbital (120 mg/kg) and the hemorrhage/reinfusion model was performed as described. 7 Briefly, the left carotid artery and the jugular vein were cannulated for blood pressure monitoring and for blood withdrawal and reinfusion, respectively. Mice were subjected to hemorrhagic shock by withdrawal of blood to allow the mean arterial blood pressure (MABP) to be maintained at 40 mmHg for 45 minutes. 7 The mean bleed out volume was 0.77±0.22 ml and 0.72±0.19 ml for the wild-type and eNOS-deficient mice, respectively. Blood was collected in a heparin-coated (5 U) syringe, and kept at 37°C until reinfusion. Mice were then resuscitated by infusion of the shed blood.
Mice were sacrificed by exsanguination 45 minutes after resuscitation, following completion of intravital microscopy studies. Control wild-type mice (sham) underwent cannulation and anesthesia for an identical period of time as the hemorrhaged mice, but were not bled. Wild-type and eNOS-deficient mice were randomly assigned to one of four experimental hemorrhage groups: (1) sham-operated wild-type mice receiving saline; (2) hemorrhage/ reinfused wild-type mice receiving saline; (3) hemorrhage/reinfused wild-type mice treated with fasudil (10 mg/kg/day ip for 3 consecutive days); and (4) hemorrhage/reinfused eNOS−/− mice treated with fasudil (10 mg/kg/day ip for 3 consecutive days). The dose and time course of fasudil treatment are based on previous publication. 22 The total number of circulating white blood cells in all experimental groups of mice was determined by a hemocytometric count of smears of blood obtained through the jugular vein cannula. The Rho-kinase inhibitor, fasudil, was provided by Asahi-Kasei Pharmaceuticals, Inc. (Shizuoka, Japan).
Intravital microscopy of mouse peri-intestinal venules
All intravital microscopy experiments were conducted in anesthetized mice, which were subjected to the hemorrhage/reinfusion protocol. Intravital microscopy was performed on mouse peri-intestinal post-capillary venules. Following exteriorization of a loop of ileal tissue via a midline laparotomy, the ileum was placed in a temperature-controlled fluid-filled Plexiglas chamber and trans-illuminated for bright-field observation of the peri-intestinal microcirculation according to a previously described procedure. 16 The ileum and mesentery were perfused throughout the experiment with a buffered K-H solution (pH 7.4, 37°C). Observations of rolling and adherent leukocytes were made with a Microphot microscope and a 40× salt water-immersion lens (Nikon Corp., Tokyo, Japan). Images were projected by a high-resolution color video camera (DC-330; DAGE-MTI, Inc., Michigan City, IN, USA) onto a high-resolution color video monitor (Multiscan 200SF; Sony), and the image recorded with a DVD recorder. All images were then analyzed using computerized imaging software (Phase 3 Image System; Media Cybernetics, Glen Mills, PA, USA) on a Pentium-based IBM-compatible computer. Red blood cell velocity was determined on-line using an optical Doppler velocimeter. 18 This method allows for the calculation of shear rates. 19
Analysis of eNOS expression in mouse mesenteric tissue
At the end of intravital microscopy, a tissue section of mesentery and intestine were rapidly removed and frozen in liquid nitrogen. Proteins were isolated from the tissues using protein lysis buffer and were separated on 7.5% SDS-PAGE and transferred to PVDF transfer membrane (Millipore). The membranes were probed with anti-eNOS monoclonal antibody (1:2500) (Transduction Laboratories, Lexington, KY, USA), anti-α-tubulin monoclonal antibody (1:5000) (Sigma Chemical Corp., St Louis, MI, USA), and total myosin light chain phosphatase (MYPT1) polyclonal antibodies (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Bands were detected with the use of the ECL detection kit (Amersham-Pharmacia Biotech, Piscataway, NJ, USA) and band intensities were quantified densitometrically using the NIH Image Program.
Rho-kinase activity assay
Rho-kinase activity in the mesenteric and intestinal tissue was measured as described with some modifications. 23 Briefly, mesenteric and intestinal tissues were homogenized in 50% fixative solution. The samples were then centrifuged at 4°C for 10 min at 1500 rpm. The pellet was washed with cold PBS and protein was extracted with urea buffer containing proteinase inhibitors. Rho-kinase phosphorylates the myosin-binding subunit (MBS) of MYPT1 at Thr853. The phosphorylation of MYPT1 was determined by immunoblotting of the protein extracts with phospho-Thr853 antibody. Rho-kinase activity was expressed as the ratio of p-MYPT1/total MYPT1.
Statistical analysis
All values are expressed as means ± standard error of the mean (SEM) of n independent experiments. Data were compared by analysis of variance (ANOVA) using post-hoc analysis with Fisher’s correct t-test. Probabilities or p-values of less than 0.05 were considered significant.
Results
Hemodynamic changes induced by hemorrhagic shock
Anesthetized wild-type mice exhibited initial MABP values in the range of 85–90 mmHg (Figure 1). In contrast, anesthetized eNOS−/− mice exhibited a moderately higher systemic blood pressure in the range of 115±10 mmHg. In control non-hemorrhaged mice, MABP values did not significantly change over the entire 90-minute observation period. In hemorrhaged mice, MABP was maintained at 40 mmHg for 45 minutes. After reinfusion of the shed blood to hemorrhaged mice, there were no significant differences in MABP among the four groups.
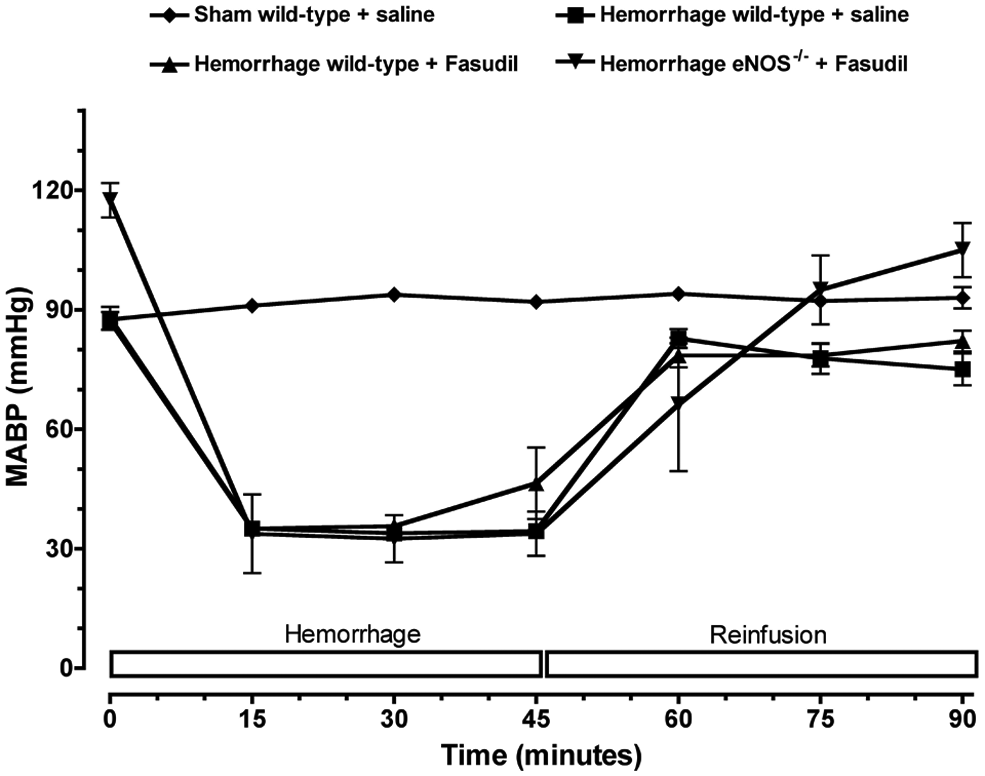
Time course of mean arterial blood pressure (MABP) during hemorrhage and reinfusion. Wild-type and eNOS-deficient mice (eNOS−/−) were subjected to hemorrhagic shock. Pharmacological inhibition of Rho-kinase was achieved by fasudil (10 mg/kg/day ip for 3 days). Each value represents mean ± SEM.
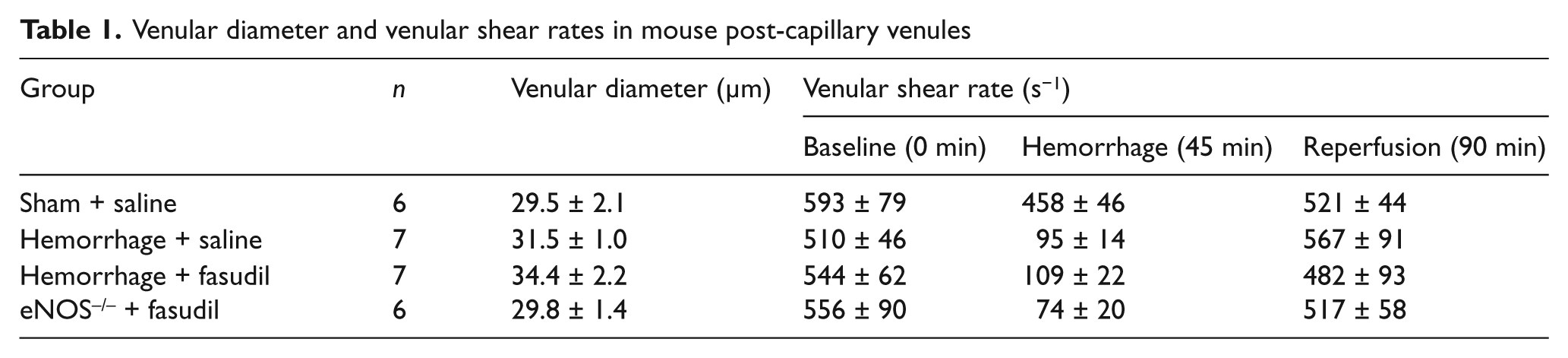
No differences were observed in venular diameter and initial shear rates among the four experimental groups of mice (Table 1). Following hemorrhage, shear rates in peri-intestinal post-capillary venules abruptly decreased to less than half of the observed initial control values or compared to sham controls. Thus, the hemorrhagic-reinfusion model is characterized by a marked hypoperfusion of the splanchnic microvasculature during the oligemic phase. However, upon reinfusion of shed blood, venular shear rates returned to normal values, which indicate that blood flow was re-established to control levels during the post-oligemic phase. Since shear rates were normal post-reinfusion, the adhesive interactions observed between leukocytes and the microvascular endothelium during resuscitation from hemorrhage could not be attributed to alterations in physical hydrodynamic forces brought about by perturbations in local hemodynamics.
Venular diameter and venular shear rates in mouse post-capillary venules
Increased leukocyte–endothelium interaction following hemorrhagic shock is mediated by Rho-kinase
A low baseline number of rolling (i.e. 25–30 cells/min) and adherent (i.e. 2–3 cells/100 µm) leukocytes were observed in the mesenteric microvasculature for all experimental groups of mice (Figures 2 and 3). Furthermore, neither the rolling nor adherence of leukocytes increased in non-hemorrhaged control mice (i.e. sham) at any time. Baseline leukocyte rolling and adherence were not significantly changed during the first 45 minutes of the hemorrhage period in wild-type and eNOS−/− mice. However, the number of rolling and adherent leukocytes in untreated hemorrhaged wild-type mice exhibited a significant increase following reinfusion compared to normal control values.
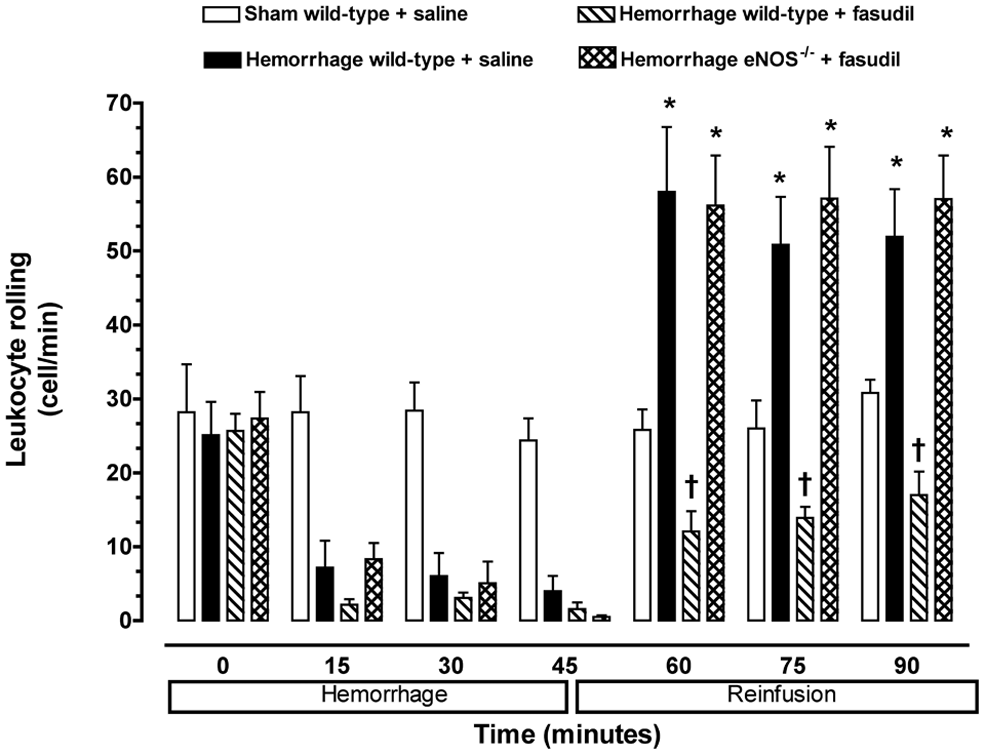
Leukocyte rolling observed in peri-intestinal post-capillary venules of wild-type mice and eNOS–\– given either saline or fasudil, and subjected to hemorrhagic shock. Values represent mean ± SEM. *p < 0.01 vs sham-operated wild-type mice; †p < 0.01 vs untreated wild-type mice and fasudil-treated eNOS−/− subjected to hemorrhage/reinfusion.
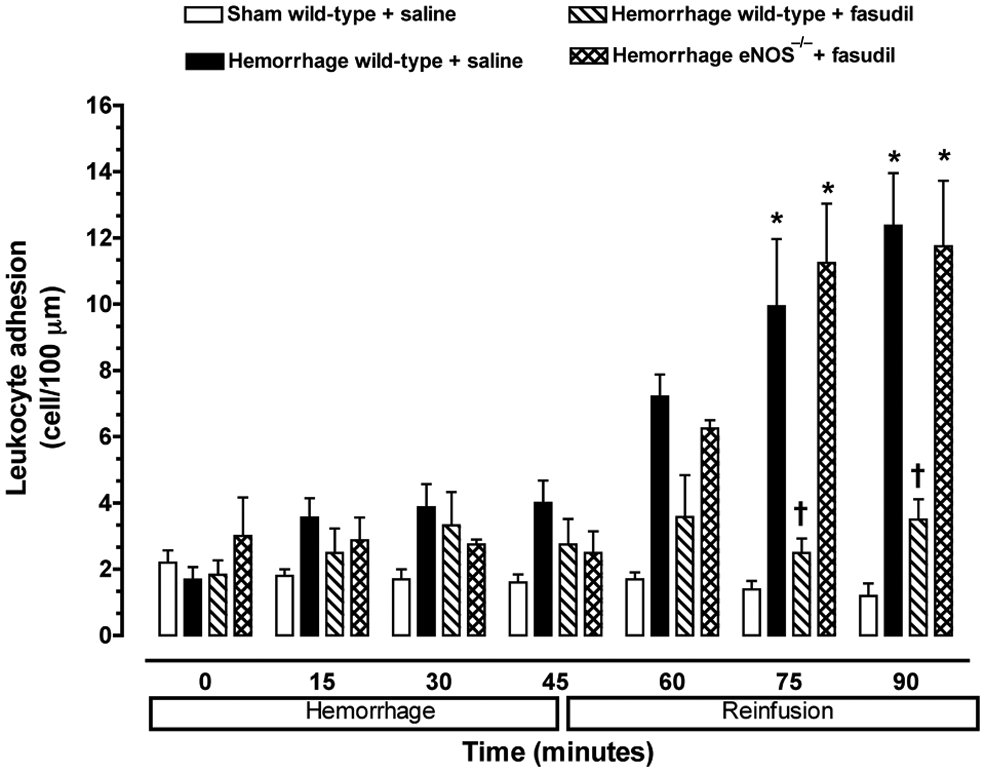
Leukocyte adhesion observed in peri-intestinal post-capillary venules of wild-type mice and eNOS–\– given either saline or fasudil, and subjected to hemorrhagic shock. Values represent mean ± SEM. *p < 0.01 vs sham-operated wild-type mice; †p < 0.01 vs untreated wild-type mice and fasudil-treated eNOS−/− subjected to hemorrhage/reinfusion.
No significant increase in the number of rolling or adherent leukocytes was observed in the peri-intestinal post-capillary venules of hemorrhaged wild-type mice treated with the Rho-kinase inhibitor fasudil (Figures 2 and 3). In contrast, fasudil did not affect leukocyte–endothelium interaction in response to hemorrhage/reinfusion in eNOS-deficient mice, suggesting that eNOS may be the main target of Rho-kinase in this model of ischemia–reperfusion injury.
No significant change in the total number of circulating leukocytes was observed between the experimental groups of mice, so that the changes in rolling and adherence could be attributed to leukopenia. The average number of circulating leukocytes in wild-type and eNOS-deficient mice was 6.0 ± 1.6 and 6.2 ± 1.4 × 103 cells/mm3, respectively. These values were not significantly different from each other, nor was leukopenia observed at the end of the experimental protocol or following systemic administration of fasudil. Therefore, Rho-kinase exerts a critical role in triggering endothelial–leukocyte interaction following hemorrhage and fluid resuscitation that is mediated, in part, by impairment of eNOS.
Evaluation of Rho-kinase and eNOS activity during ischemia–reperfusion injury
Rho-kinase activity was not affected by the loss of eNOS as phosphorylation of MYPT1 was similar between wild-type and eNOS−/− mice (Figure 4). Intraperitoneal administration of fasudil to wild-type mice, however, inhibited Rho-kinase activity in mesenteric and intestinal tissues. The reduction in Rho-kinase activity correlated with the attenuation in leukocyte–endothelium interactions. Similarly, treatment with fasudil significantly reduced Rho-kinase activity in eNOS−/− mice to a level comparable to that observed in wild-type mice.
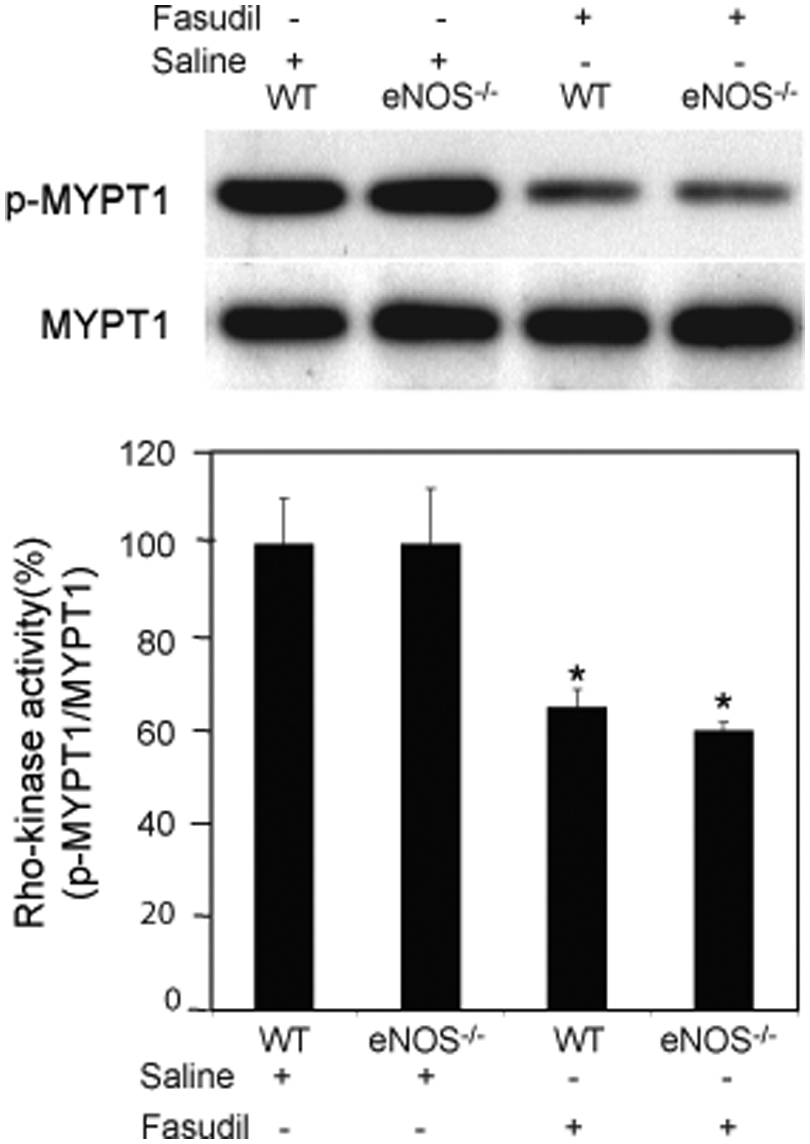
Rho-kinase activity as measured by phosphorylation of MYPT1 in wild-type and eNOS−/− mice. Wild-type and eNOS−/− mice were treated with either saline or fasudil. Protein was extracted from mesenteric and intestinal tissues. Rho-kinase activity was expressed as the ratio of p-MYPT1/total MYPT1. Values represent mean ± SEM. *p < 0.01 vs untreated mice.
To test the hypothesis that inhibition of Rho-kinase attenuates the inflammatory response in an eNOS-dependent manner, we analyzed the eNOS expression level in wild-type mice treated with fasudil. Expression of eNOS (normalized to α-tubulin) was upregulated in wild-type mice treated with fasudil for 3 days (Figure 5). To determine the role of eNOS in mediating the inhibitory effects of fasudil on the leukocyte–endothelium interaction during hemorrhage/reinfusion, we studied leukocyte–endothelium interactions in eNOS−/− mice treated with fasudil. In contrast to wild-type mice, fasudil treatment failed to inhibit hemorrhage-induced leukocyte rolling and adherence in eNOS−/− mice (Figures 2 and 3), despite similar Rho-kinase inhibition in wild-type mice (Figure 4). These findings strongly suggest that upregulation of eNOS contributes to the inhibition of endothelial–leukocyte interactions by fasudil following resuscitation from hemorrhagic shock.
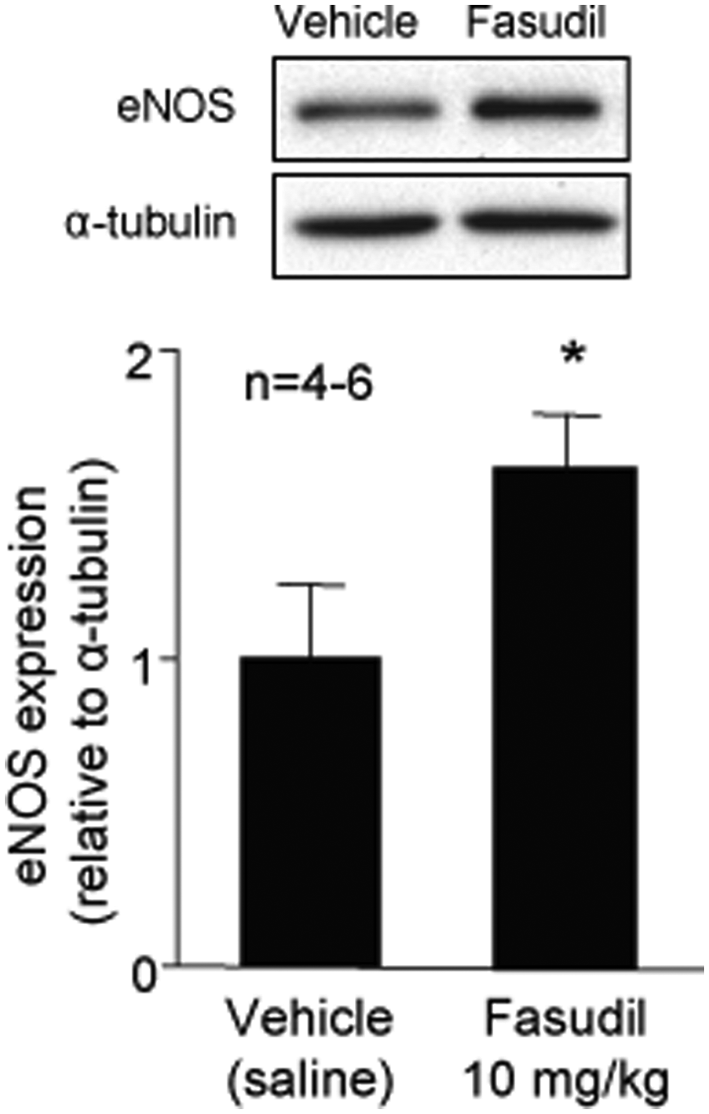
eNOS protein levels after fasudil treatment. Wild-type mice were treated with fasudil (10 mg/kg/day ip for 3 days). Protein levels of eNOS were normalized to α-tubulin. Values represent mean ± SEM. *p < 0.01 vs untreated mice.
Discussion
This study was undertaken to determine the mechanisms of Rho-kinase in regulating the early inflammatory response after resuscitation from hemorrhage. Evidence shows that Rho-kinase activity is increased following ischemia–reperfusion injury 24 and activation of Rho-kinase promotes leukocyte infiltration. 17 The detrimental inflammatory response has also been shown to be triggered by loss of eNOS due to endothelial dysfunction during reperfusion injury. However, it is not known if Rho-kinase regulates leukocyte recruitment during ischemia–reperfusion injury via the eNOS pathway. We showed that inhibition of Rho-kinase upregulates eNOS expression in vivo, which correlates with attenuation in leukocyte–endothelium interactions. In addition, the vascular protective effects of the Rho-kinase inhibitor were absent in eNOS−/− mice. These findings indicate that eNOS expression is negatively regulated by Rho-kinase and that the downregulation of eNOS plays a key role in the activation of the inflammatory cascade that occurs after ischemia–reperfusion injury. The Rho-kinase/eNOS pathway, therefore, may serve as a therapeutic target for reducing the inflammatory response following ischemia–reperfusion injury.
Resuscitation from hemorrhagic shock represents a severe form of whole-body ischemia–reperfusion injury in which a multiple series of adhesive and signaling events regulate the activation of inflammatory responses.25,26 Several studies suggest that a common pathophysiologic event occurring during ischemia–reperfusion is the early development of acute endothelial dysfunction characterized by impaired release of endothelium-derived NO.27–29 Indeed, endothelial dysfunction associated with severe organ injury has been reported in myocardial 30 and splanchnic 31 ischemia–reperfusion injury, and traumatic 11 and hemorrhagic shock. 10 In addition, endothelial dysfunction occurs in the mesenteric vasculature (i.e. 15–30 minutes) following ischemia–reperfusion injury.10,32 Endothelial dysfunction is characterized by impaired vasorelaxation in response to endothelium-dependent vasodilators due to diminished release or bioavailability of endothelium-derived NO. 32 This decrease in the release of endothelium-derived NO facilitates increased neutrophil adhesion3,33 and transendothelial migration through increased expression of endothelial adhesion molecules (i.e. P-selectin and ICAM-1), a process that normally is suppressed by physiologic amounts of endothelium-derived NO. 34 The overall consequence of endothelial dysfunction, therefore, is the infiltration and activation of neutrophils in ischemic tissues.2,11 Activated neutrophils could, in turn, release cytotoxic mediators that could lead to further microcirculatory dysfunction that is associated with fluid loss and further neutrophil infiltration into the injured tissues. 7 This process could ultimately lead to marked hypotension and end-organ damage.
In the present study, we found that after resuscitation from hemorrhagic shock, endothelial–leukocyte interactions are substantially augmented in the microcirculation, an event that is associated with increased Rho-associated coiled-coil-containing kinases (ROCKs) activity and impaired eNOS expression. Interestingly, the increase in endothelial–leukocyte interactions was prevented by treatment with the Rho-kinase inhibitor, fasudil. This is consistent with previous studies showing that inhibition of Rho-kinase increases eNOS mRNA stability, 18 and can lead to the rapid activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway, which can phosphorylate and activate eNOS. 35 Indeed, our data showed that Rho-kinase inhibition leads to increased eNOS protein levels and activity (data not shown). Because inhibition of Rho-GTPase by statins could also protect the cardiovascular system against reperfusion injury, it is likely that the beneficial effects of Rho-GTPase inhibition are attributed to the inhibition of its downstream target, Rho-kinase. Indeed, statins, which are known to inhibit Rho-GTPases, increase the half-life and activity of eNOS, both in vitro 36 and in vivo. 37 The inhibition of Rho-GTPases by statins involve the blocking of the geranylgeranyl-pyrophosphate (GGPP)-dependent isoprenylation of Rho, which leads to the inhibition of Rho activation 18 and reduced activity of its downstream target, Rho-kinase. 19 Our findings are also consistent with an earlier report demonstrating that inhibition of Rho-kinase activity markedly reduced tissue injury in response to ischemia, 19 although the effect on leukocyte–endothelium interactions was not demonstrated in earlier studies.
The inhibitory effect on endothelial–leukocyte interaction exerted by inhibition of Rho-kinase may contribute to normalization of the pathophysiologic events during hemorrhage/reinfusion injury through several mechanisms. One possibility is that preservation of eNOS by Rho-kinase inhibition may block early tethering of leukocytes to the endothelium, thus diminishing the local production of pro-inflammatory cytokines, which could induce expression of endothelial cell adhesion molecules. Indeed, the loss of endothelial-derived NO leads to the rapid increase in P-selectin expression at the endothelial cell surface, which orchestrates the initial early onset of leukocyte–endothelium interaction during inflammation.27,33,38 Moreover, a functional relationship between loss of endothelium-derived NO and the upregulation of P-selectin on the venular endothelium has been previously reported. 31 Therefore, ischemia-induced loss of endothelium-derived NO release is likely responsible for increased expression of cellular adhesion molecules in the microvascular endothelium. This is consistent with previous observations that endothelial dysfunction occurs very early following hemorrhagic shock and persists despite fluid resuscitation, 39 and that a monoclonal antibody against P-selectin could attenuate the outcome of hemorrhagic shock. 7 Overall, inhibition of leukocyte infiltration into the vessel wall exerts a key anti-inflammatory effect because activated neutrophils, which have adhered to the endothelium, could release cytotoxic mediators including proteases, eicosanoids, cytokines, and oxygen-derived free radicals,2,11,12 each of which can promote further tissue injury and exacerbate endothelial dysfunction in ischemic organs.
The role of Rho-kinase in pathophysiology after hemorrhagic shock has also been demonstrated in regulating vascular reactivity.40–43 After hemorrhagic shock, vascular responses to vasoconstrictor and vasodilators display a biphasic pattern with hyperactivity occurring immediately after shock and hyporeactivity occurring at a later phase.40,43 Interestingly, Rho-kinase activity increases during the early phase of hemorrhagic shock and its activity tends to diminish during the later phases of shock. 40 In addition, angiotensin II, an activator of Rho-kinase, improves calcium sensitivity and hemorrhagic shock-induced hyporeactivity. 40 However, the role of Rho-kinase in vascular reactivity and inflammation during reperfusion is unclear. Our data suggest that inhibition of Rho-kinase during reperfusion generates a beneficial effect by restoring endothelial function and reducing inflammation. These findings suggest that Rho-kinase inhibitors may have therapeutic benefits in ischemic cardiovascular diseases.
Besides regulating eNOS, ROCK has been shown to activate Jun N-terminal kinases (JNKs) in response to inflammatory cytokines.44,45 Inhibition of ROCK attenuated TNF-α or lysophosphatidic acid (LPA)-induced JNK activation. In addition, activation of Rho-kinase is required for JNK-dependent interleukin (IL)-6 secretion induced by TNF-α. It is possible that ROCK may modulate inflammation during reperfusion via the JNK pathway in addition to eNOS regulation. On the other hand, there is also evidence suggesting that eNOS regulates JNK activation. Pre-treatment with the eNOS inhibitor (L-NAME) reduced hypoxia-induced JNK activation in endothelial cells. 46 However, the mechanism of eNOS-mediated JNK activation is not clear.
In summary, we have shown that inhibition of Rho-kinase during the reperfusion phase of hemorrhagic shock leads to decreased endothelial–leukocyte interaction via an eNOS-dependent mechanism. These findings suggest that Rho-kinase inhibitors may have therapeutic benefits in the phase of reperfusion after hemorrhagic shock by preserving endothelial function, limiting the infiltration of leukocytes, and reducing vascular inflammation.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health (HL052233, HL091933, NS070001, DK085006).
Conflict of interest statement
JK Liao is a consultant for Asahi-Kasei Pharmaceuticals, Inc. All of the other authors have no declaration of conflicts of interests.