
Abstract
Patients with fibromuscular dysplasia (FMD) may have clinical features consistent with Mendelian vascular connective tissue disorders. The yield of genetic testing for these disorders among patients with FMD has not been determined. A total of 216 consecutive patients with FMD were identified. Clinical characteristics were collected and genetic test results reviewed for abnormalities in the following genes: transforming growth factor-β receptor 1 and 2 (TGFβR1 and TGFβR2), collagen 3A1, fibrillin-1, smooth muscle α-actin 2, and SMAD3. A total of 63 patients (63/216; 29.2%) were referred for genetic counseling with testing performed in 35 (35/63; 55.6%). The percentage of patients with a history of arterial or aortic dissection, history of aortic aneurysm, systemic features of a connective tissue disorder, and a family history of sudden death was significantly larger in the group that underwent genetic testing (62.9% vs 18.2%, p < 0.001; 8.6% vs 1.7%, p = 0.02; 51.4% vs 17.1%, p < 0.001; and 42.9% vs 22.7%, p = 0.04, respectively). Two patients were found to have distinct variants in the TGFβR1 gene (c.611 C>T, p.Thr204lle and c.1285 T>C, p.Tyr429His). The yield of genetic testing for vascular connective tissue disorders was low in a high-risk subset of FMD patients. However, two patients with a similar phenotype had novel and distinct variants in the TGFβR1 gene, a finding which merits further investigation.
Introduction
Fibromuscular dysplasia (FMD) is an uncommon, non-inflammatory arterial disease that primarily occurs in women. There is a tendency for involvement of medium-sized arteries with the renal, carotid, and vertebral arteries most commonly affected. 1 FMD is traditionally classified into categories based on the layer of the arterial wall in which the lesion is localized. 2 Medial fibroplasia is the most common type of FMD2,3 and has a classic ‘string of beads’ appearance on angiography. Intimal fibroplasia occurs in less than 10% of patients with FMD and results in the angiographic findings of focal concentric stenosis or tubular lesions.2,3 Perimedial fibroplasia, which is rare, involves fibrosis on the outer aspect of the medial layer and has an angiographic appearance of limited beading with beads that are smaller than the vessel lumen.3,4 Adventitial fibroplasia is the rarest type of lesion2,3 with angiographic images similar to those of intimal disease. 4 Classification of FMD was historically determined by histology; however, in the modern era of endovascular therapy, patients rarely undergo open surgical procedures. As such, FMD type is classified according to angiographic appearance of the vascular lesions which have been validated with histopathological–angiographic correlation studies.4,5
The presence of FMD in an arterial bed may lead to arterial stenosis, dissection, or aneurysm. Clinically, patients with renal artery FMD often present with hypertension. 6 In patients with carotid or vertebral artery FMD, symptoms and signs may include neck pain, pulsatile tinnitus, Horner’s syndrome due to carotid artery dissection, transient ischemic attack (TIA), and cerebral infarction. 6 Alternatively, FMD may be asymptomatic and detected by imaging performed for another reason or in evaluation of a vascular bruit heard on physical examination.
Although multiple theories have been proposed, including genetic and environmental factors, such as estrogen exposure and tobacco use, 7 the etiology of FMD is unknown. A number of case reports describe FMD in siblings8–12 and first degree cousins 13 and support a genetic contribution to the pathogenesis of the disease. The first formal genetic analysis was conducted by Rushton and colleagues in a study of 20 families in which at least one member had a diagnosis of FMD. The findings of this study suggested an autosomal dominant trait with variable penetrance in 60% of the families.14,15 Similarly, Mettinger and Ericson examined the pedigrees of 37 patients with cerebrovascular FMD and concluded a dominant trait with reduced penetrance. 16 A major limitation of both of these studies was that the diagnosis of FMD in family members was based on non-specific clinical features and was not angiographically determined. Subsequent studies used imaging to diagnose familial FMD and again proposed an autosomal dominant mode of inheritance.17,18 In a study conducted by Pannier-Moreau and colleagues, an 11% prevalence of familial FMD was found in the renal arteries. 19
To date, there has been no single genetic abnormality rigorously associated with FMD. There have been a number of reports connecting FMD with α1-antitrypsin deficiency.20–23 In addition, a single study found that FMD patients had a significantly higher frequency of the angiotensin-converting enzyme (ACE) I allele, which has been associated with decreased levels of circulating ACE. 24 Finally, Sang and colleagues found a higher prevalence of the class II human lymphocytic antigen (HLA)-DRw6 in patients with FMD compared to control subjects. 7
Further studies provide evidence for an overlap of FMD with vascular connective tissue disorders that have known Mendelian genetic abnormalities. Ehlers Danlos syndrome type IV (EDS type IV, also known as vascular EDS) is caused by mutations in the gene for type III procollagen (COL3A1), and two separate reports describe coexistence of FMD and EDS type IV in a single patient.25,26 Schievink and colleagues also report both FMD and cystic medial necrosis in a young female patient with Marfan syndrome, which is caused by mutations in fibrillin-1 (FBN1). 27 Although not previously described, additional connective tissue disorders that may be considered in patients with FMD, depending upon clinical presentation and imaging findings, include Loeys-Dietz syndrome, aneurysms-osteoarthritis syndrome, and arteriopathy associated with mutations in the smooth muscle α-actin 2 (ACTA2) gene.
Loeys-Dietz syndrome is characterized by arterial tortuosity with arterial or aortic aneurysm and/or dissection, hypertelorism, and bifid uvula or cleft palate.28,29 The phenotype is caused by mutations in the genes encoding transforming growth factor-β receptors 1 and 2 (TGFβR1 and TGFβR2, respectively).28,29 Aneurysms-osteoarthritis syndrome is similarly characterized by arterial pathology as well as early onset osteoarthritis.30,31 This syndrome has been linked to mutations in SMAD3, which similarly to those in TGFβR1 and TGFβR2, lead to increased TGF-β signal transmission.30,31 SMAD3 mutations have also been described in individuals with arterial aneurysm and/or dissection in whom additional features of the aneurysms-osteoarthritis syndrome are absent. 32 Finally, mutations in ACTA2 were originally described as causing a non-syndromic form of familial thoracic aortic aneurysm and dissection.33,34 However, further investigation revealed a role for these mutations in a wide spectrum of arteriopathy including coronary artery and Moyamoya disease. 35
Patients with FMD and clinical features consistent with a known vascular connective disorder may be referred for genetic counseling and subsequent performance of genetic testing. Suspicious clinical characteristics might include arterial aneurysm and/or dissection, marked arterial tortuosity, a family history of aneurysm, dissection, or sudden death, or physical examination findings such as hypermobility, deformities of the chest wall (pectus excavatum or pectus carinatum) or uvula, a high-arched palate, or skin findings such as atrophic scars. The clinical utility of such genetic testing in a population of FMD patients is unknown. The objective of this study was to determine the yield of testing for known vascular connective tissue disorders among patients with FMD referred for genetic testing. An additional goal was to determine the clinical characteristics of those patients who underwent genetic testing.
Methods
Patients evaluated in the Cleveland Clinic FMD program were identified from a single center database of the United States FMD Patient Registry. 36 All patients included in the database provided informed consent and had a diagnosis of FMD that was confirmed by imaging (arteriography, computed tomographic angiography, magnetic resonance angiography, or duplex ultrasound examination) reviewed by a physician staff member of the FMD program. Additional clinical characteristics were collected for each patient through review of the single-center database as well as added review of the electronic medical record. Clinical features included history of arterial or aortic dissection, history of arterial or aortic aneurysm, history of TIA or stroke, systemic findings of a connective tissue disorder, and family history. Patients considered to have systemic findings of a connective tissue disorder had one or more of the following documented in their electronic medical record: Marfanoid body habitus (tall stature, reduced upper segment to lower segment ratio, and increased arm to height ratio), positive wrist sign, positive thumb sign, pectus carinatum, pectus excavatum, scoliosis, dysmorphic facial features, joint hypermobility, atrophic scars, pinched nasal bridge, translucent and/or velvety skin, high-arched palate, uvula deformity, hypertelorism, livedo reticularis, dental crowding, pes cavus or planus, mandibular torus, and arachnodactyly.
In addition to clinical characteristics, information regarding referral to a genetic counselor and performance of genetic testing for known vascular connective tissue disorders was obtained. Specifically, genetic test results were reviewed for abnormalities in the following genes: TGFβR1, TGFβR2, COL3A1, FBN1, ACTA2, and SMAD3. Results of genetic testing performed for non-vascular indications were also documented, if available. Among those who underwent genetic testing, samples were obtained from a peripheral blood draw and analyzed in one or more of the following laboratories, all of which were Clinical Laboratory Improvement Amendments (CLIA) accredited: Connective Tissue Gene Tests (Allentown, PA, USA), Collagen Diagnostic Laboratory (University of Washington, Seattle, WA, USA), and Laboratory for Molecular Medicine, Center for Genetics and Genomics (Harvard Medical School, Cambridge, MA, USA). At each laboratory, all exons in the gene of interest were amplified by polymerase chain reaction. The amplified products were then sequenced using an automated sequencer and analyzed for variations.
Statistical analysis was performed using SPSS software (IBM Corporation, Armonk, NY, USA). Descriptive statistics are presented as means ± standard deviations, percentages, and frequencies. A chi-square analysis was used to compare the clinical characteristics of those who underwent genetic testing versus those who were not tested. A P-value ≤ 0.05 was considered statistically significant. All research was conducted in accordance with the Institutional Review Board at the Cleveland Clinic.
Results
A total of 216 consecutive FMD patients were identified. Patient demographics and FMD type are shown in Table 1. Mean age at the time of diagnosis was 48.7 ± 13.5 years. Ninety-four percent of patients were female and 94.9% were Caucasian. Medial fibroplasia was the most common type of FMD and was present in 84.7% of patients. Intimal fibroplasia was diagnosed less frequently in 6.5% of patients. No patients had perimedial fibroplasia and in 8.8% the type of FMD could not be definitively determined with available imaging.
Patient demographics and FMD type
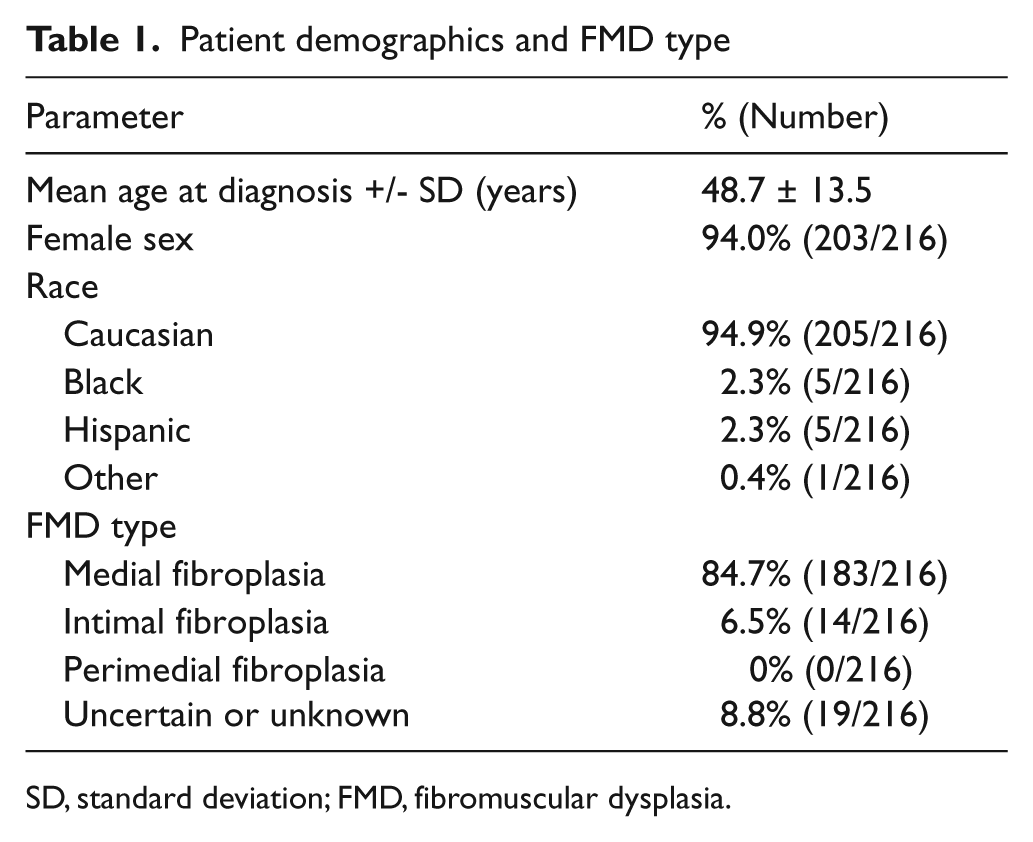
SD, standard deviation; FMD, fibromuscular dysplasia.
Vascular bed involvement is depicted in Table 2. Given both the retrospective nature of our study and the lack or consensus practice guidelines as to the optimal management of patients with FMD, patients were not screened for potential involvement of all vascular beds. In most patients, the renal and extracranial carotid arteries were imaged, with imaging of additional vascular beds performed in cases of high clinical suspicion (e.g. symptoms or physical examination findings). The extracranial carotid and/or vertebral arteries were most commonly affected, with imaging-confirmed FMD present in 81.0% of patients. FMD involving the renal arteries was also frequent and found in 61.6% of patients. Lesions affecting the intracranial (including cerebral artery aneurysms), mesenteric, and lower extremity arteries were diagnosed in 13.4%, 14.8%, and 15.7% of patients, respectively. Involvement of the upper extremity arteries, coronary arteries, and aorta was also found, however, in only a small number of patients. The single patient with aortic involvement was diagnosed with FMD at age 5 years after presenting with middle aortic syndrome, inferior mesenteric artery aneurysm, and celiac, superior mesenteric, and bilateral renal artery stenosis. A diagnosis of Takayasu arteritis had been excluded in this patient.
Vascular bed involvement
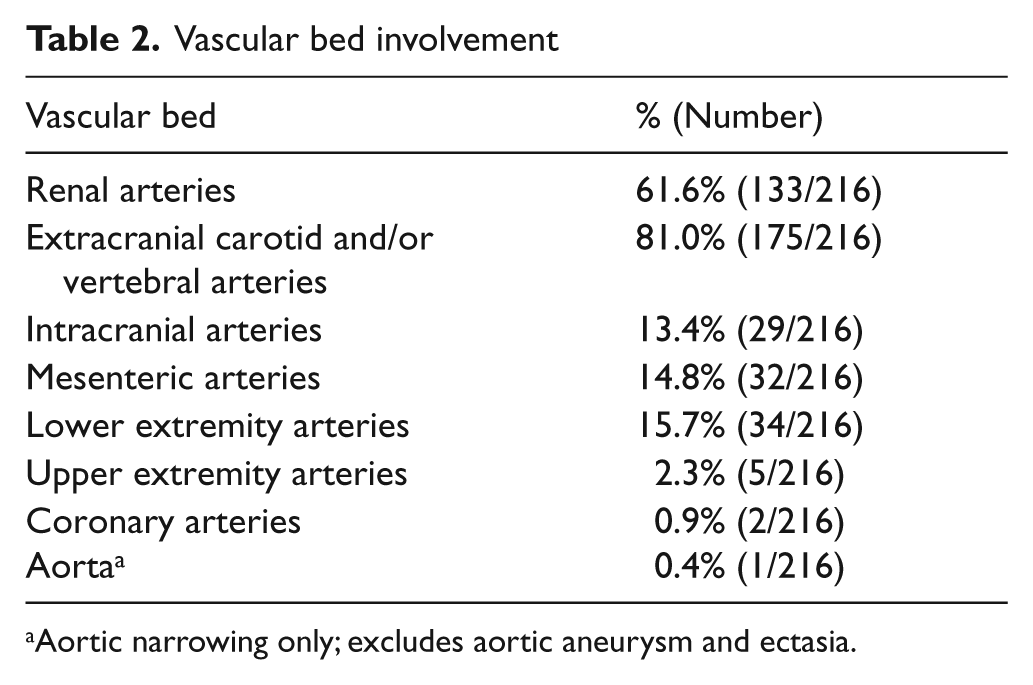
Aortic narrowing only; excludes aortic aneurysm and ectasia.
A total of 63 patients (63/216; 29.2%) were referred for genetic counseling (Table 3). Genetic counseling only (no testing) was performed in 44.4% of these patients (28/63) or in 13.0% of the total patient population (28/216). Genetic testing was performed in 55.6% of those referred for counseling (35/63) or in 16.2% of the total patient population (35/216). Among those who were referred for counseling but did not undergo testing, 46.4% (13/28) were not offered testing due to a low likelihood of identifying a vascular connective tissue disorder. In 14 of the remaining patients, genetic testing for at least one vascular connective tissue disorder was offered. Four of these patients declined genetic testing, one was unable to pursue testing for financial reasons, five requested additional time to consider testing, and four agreed to be tested elsewhere under the supervision of their primary care provider. The final patient was a child with renal FMD and aortic insufficiency, and genetic testing was recommended for his father who had a history of a bicuspid aortic valve. TGFβR1, TGFβR2, and COL3A1 were the genes most frequently investigated, with testing performed in 85.7%, 85.7%, and 80.0% of patients who underwent genetic testing, respectively (Table 3). In addition, 28.6% of patients were tested for mutations in ACTA2, 5.7% for mutations in FBN1, and 8.6% for mutations in SMAD3.
Summary of genetic counseling and testing

The clinical characteristics of patients who underwent genetic testing versus those who were not tested are shown in Table 4. There were no differences between the groups in regard to the type of FMD. The percentage of patients with involvement of the intracranial arteries was significantly larger in the group that underwent genetic testing (25.7% vs 11.0%, p = 0.001). Although not statistically significant, there was a larger percentage of patients with involvement of the extracranial carotid and/or vertebral arteries in those who underwent testing (91.4% vs 79.0%). As expected, the percentage of patients with a history of arterial or aortic dissection, history of aortic aneurysm, systemic features of a connective tissue disorder, and a family history of sudden death was significantly larger in the group who underwent genetic testing (62.9% vs 18.2%, p < 0.001; 8.6% vs 1.7%, p = 0.02; 51.4% vs 17.1%, p < 0.001; and 42.9% vs 22.7%, p = 0.04, respectively). Although not statistically significant, there was a larger percentage of patients with a history of peripheral (i.e. non-aortic) arterial aneurysm, TIA or stroke, and a family history of arterial aneurysm in the group that underwent genetic testing (25.7% vs 14.9%, 37.1% vs 23.8%, and 40.0% vs 22.7%, respectively). There were negligible differences between groups in regard to family history of FMD, family history of dissection, or family history of stroke.
Clinical characteristics of patients who underwent genetic testing versus those who were not tested
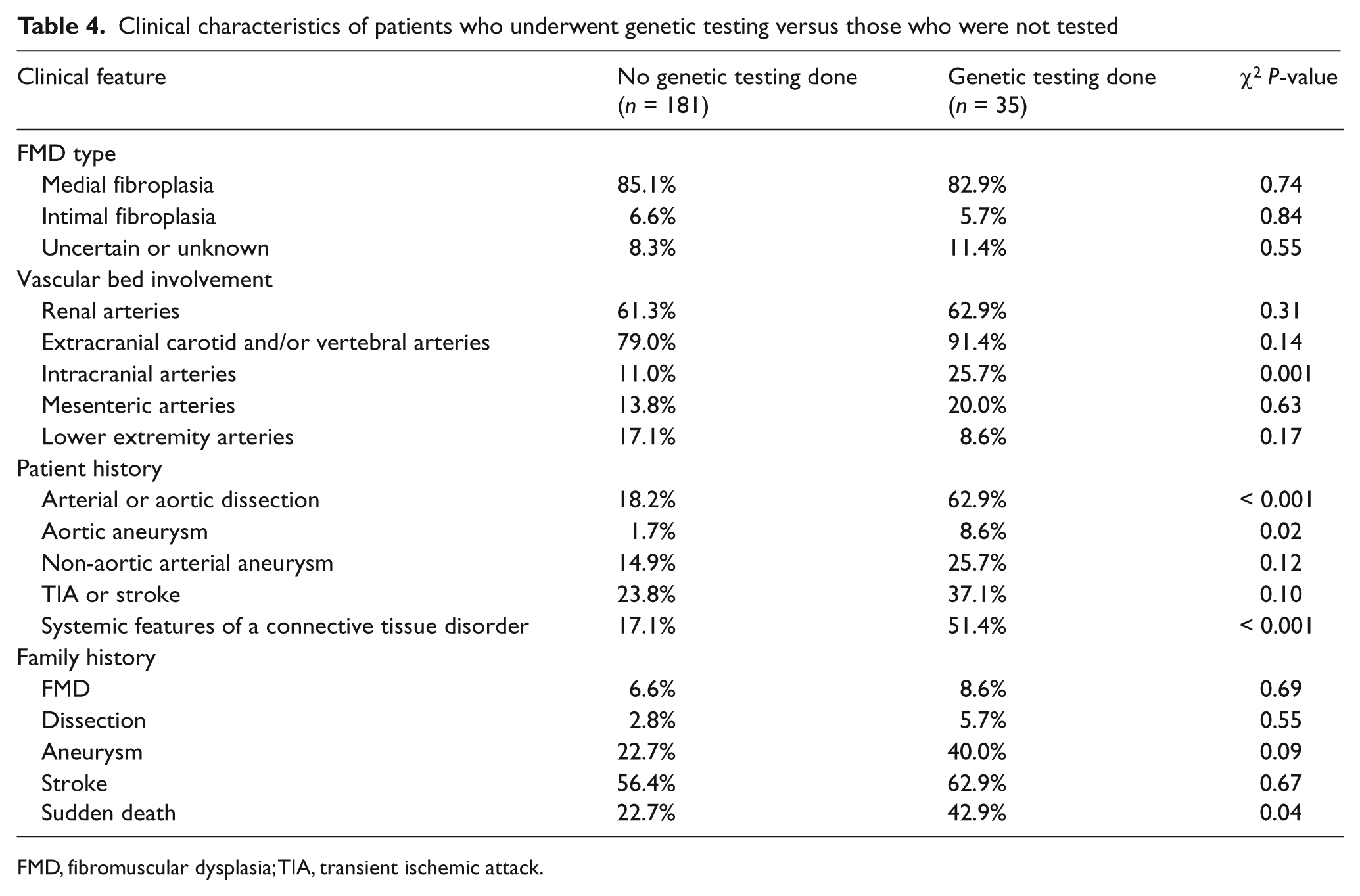
FMD, fibromuscular dysplasia; TIA, transient ischemic attack.
Results of genetic testing
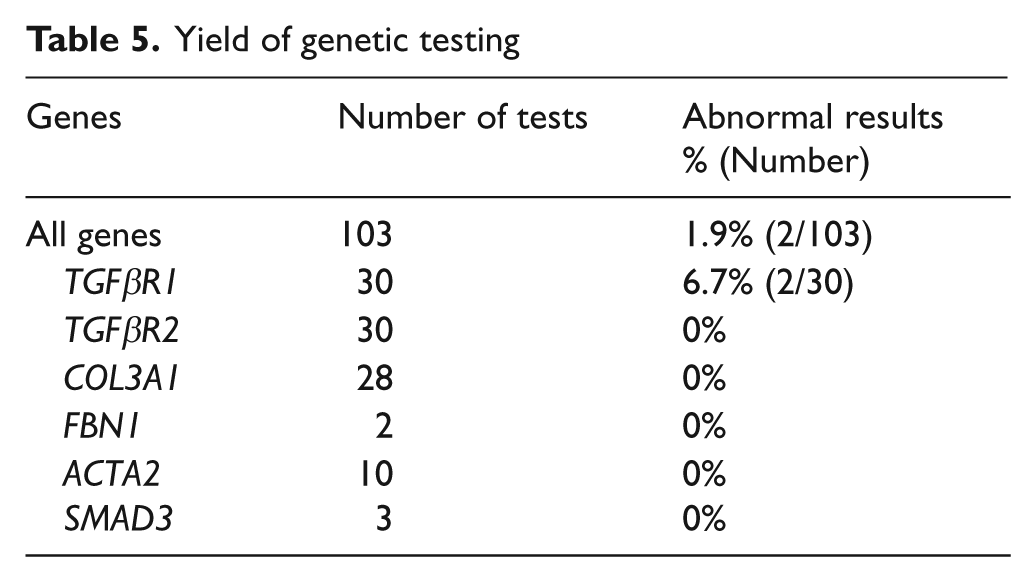
In the 35 patients tested for genetic abnormalities, a total of 103 tests were performed (Table 5). There were no mutations identified in the TGFβR2, COL3A1, FBN1, ACTA2, or SMAD3 genes. Two distinct variants were identified in the TGFβR1 gene in two separate patients (c.611 C>T, p.Thr204lle and c.1285 T>C, p.Tyr429His). Both patients were female and diagnosed with medial fibroplasia. In addition, both had a history of multi-vessel dissection involving the internal carotid and vertebral arteries and ectasia or aneurysm of the ascending aorta (Figure 1). Patient 1 reported sudden death in her father at the age of 47 years from an uncertain cause. Patient 2 reported sudden death in her father at the age of 57 years. Similarly, the cause of death was unknown. Neither patient had gross craniofacial abnormalities. Both patients had a unifid uvula and a normal appearing palate, although in one patient a short uvula was documented.
Yield of genetic testing
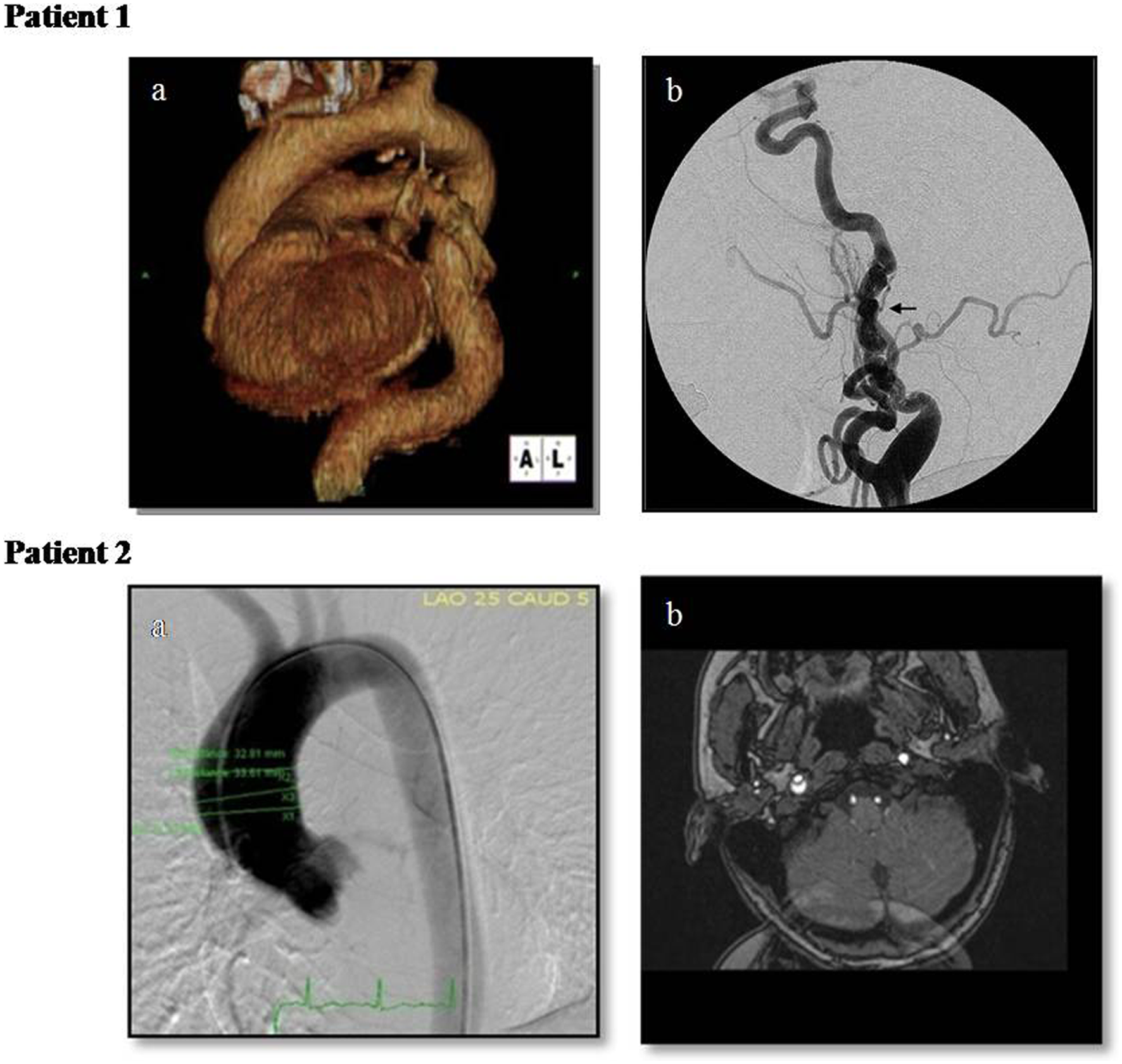
Patient 1: (a) Computed tomographic angiography showing ascending aortic aneurysm and marked tortuosity of the descending aorta; (b) angiogram of the left internal carotid artery showing tortuosity and beaded appearance consistent with medial fibroplasia (FMD). Patient 2: (a) Aortogram showing ectasia of the ascending aorta; (b) magnetic resonance angiography showing dissection of the right internal carotid artery.
Although not tested for vascular indications, a number of patients were found to have other genetic abnormalities during evaluation of comorbid medical issues. A single patient with intimal fibroplasia involving the right renal and left internal carotid arteries and prior aneurysm of the left internal carotid artery had genetic testing diagnostic of hereditary multiple exostosis and hereditary non-polyposis colorectal cancer. A second patient with medial fibroplasia involving bilateral renal and internal carotid arteries was found to have heterozygosity for the DF508 mutation associated with cystic fibrosis. A third patient with medial fibroplasia involving both renal arteries and history of right renal artery dissection had a heterozygous mutation in one of the genes associated with Familial Mediterranean fever. Hereditary non-polyposis colorectal cancer, cystic fibrosis carrier status, and Familial Mediterranean fever carrier status were believed to be unrelated to coexistent vascular disease. One pediatric patient with multi-vessel FMD, initially diagnosed at age 3 years after presenting with hypertension and renal artery stenosis, underwent testing for neurofibromatosis at an outside facility with no genetic mutation identified.
Discussion
FMD is an uncommon arterial disease, the cause of which remains unknown. A role for genetics in the pathogenesis is supported by a number of studies identifying FMD in family members,8–13,17–19 association of FMD with a variety of genetic abnormalities,7,20–24 and the coexistence of FMD with Mendelian vascular connective tissue disorders.25–27 Patients with FMD and a clinical phenotype consistent with these disorders may be referred for genetic counseling and eventual testing; however, the diagnostic yield of genetic testing for Mendelian connective tissue disorders in this population has not been determined. We identified 216 consecutive, well-phenotyped patients, with a confirmed diagnosis of FMD seen in the FMD program of the Cleveland Clinic from a single-center database of the United States FMD Patient Registry. The registry database and electronic medical record were reviewed for collection of clinical characteristics and information regarding genetic testing. Our series of FMD patients had a prevalence of cerebrovascular (extracranial carotid and/or vertebral artery) and intracranial carotid artery involvement (81.0% and 13.4%, respectively) that was similar to findings of the recent publication by the United States Registry for FMD, which reported extracranial carotid artery involvement in 74.3% of patients and intracranial carotid artery involvement in 17.0%. 36 It should be noted, that there is significant overlap of our study population and the United States Registry for FMD, as our single-center registry data were used to identify and characterize FMD patients included in this study. Cleveland Clinic has been an active participant in the United States Registry for FMD. Of note, both our series and that of the United States Registry had a significantly higher prevalence of cerebrovascular involvement than has been previously reported in the literature, 1 a fact which likely reflects the quaternary nature and unique referral patterns of our center as well as others involved in the registry.
Thirty-five patients underwent genetic testing for mutations in at least one of the following genes: TGFβR1, TGFβR2, COL3A1, FBN1, ACTA2, and SMAD3. This was a high-risk subset of FMD patients with a high prevalence of intracranial (including cerebral aneurysms) involvement, arterial or aortic dissection, aortic aneurysm, systemic features of a connective tissue disorder, and a family history of sudden death. The overall yield of genetic testing was low, with only two potential abnormalities identified out of a total of 103 tests performed (1.9%). The variants occurred in two separate patients with similar clinical phenotypes, and both variants were located in the TGFβR1 gene (6.7% of patients tested for abnormalities in this gene: c.611 C>T, p.Thr204lle and c.1285 T>C, p.Tyr429His). Both patients experienced multiple cervical artery dissections and had ectasia or aneurysm of the ascending thoracic aorta in addition to classic medial FMD lesions.
Literature that describes genetic testing for mutations associated with vascular connective tissue disorders in patients with FMD is sparse. An abstract presented by McDonnell and colleagues at the American Society of Human Genetics Annual Meeting in 2006 described a cohort of 30 patients with FMD and features of EDS type IV. 37 However, similar to our study, there were no patients with abnormalities in COL3A1. The authors additionally reported no abnormalities in the TGFβR1 and TGFβR2 genes. The phenotype of this cohort consisted of arterial aneurysms and dissections, atrophic scars, velvety or stretchy skin, joint hypermobility, articular dislocation, uterine prolapse, chest wall deformities, pes planus, and scoliosis, and the authors proposed a previously unrecognized form of EDS, distinct from EDS type IV, with FMD as a major clinical feature. 37 A second study performed sequencing of the ACTA2 gene in 13 pediatric patients with histologically or angiographically confirmed FMD and similarly reported no abnormalities. 38
Novel variants in the TGFβR1 gene
TGFβR1
The gene encoding TGFβR1 is located on the long arm of chromosome nine at position 22.33 (9q22.33). 39 It is approximately 31 kilobases long and contains nine exons. 40 The gene encodes an extracellular domain, a transmembrane domain, and a large intracellular domain composed of the serine-threonine kinase domain (catalytic) and glycine-serine-rich domain (contains sites of activating phosphorylation). Through binding of its ligand, TGF-β, the receptor is thought to play a role in cell-cycle progression. 41
c.611 C>T, p.Thr204lle
Patient 1 (Figure 1) was found to have a heterozygous variant of unknown significance in exon 4 at position 204 in which threonine was replaced with isoleucine. Position 204 is located at the end of the junction between the glycine-serine-rich domain and the serine-threonine kinase domain. At present, no function has been assigned to the junction region. However, threonine at position 204 of the TGFβR1 is highly conserved across species, which may indicate functional significance. Information regarding familial segregation is not available for this patient, as family members declined additional testing.
Although a number of mutations have been identified in the TGFβR1 gene,28,29,42 there are no previous reports of this specific variant. However, in their original description of Loeys-Dietz syndrome, Loeys et al. reported an identical substitution within the junction region at position 200. 28 The patient was a 15-month-old male with a phenotype characterized by aortic root and pulmonary artery aneurysms, hypertelorism, cleft palate, bifid/broad uvula, pectus deformity, scoliosis, and joint laxity. 28
c.1285 T>C, p.Tyr429His
Patient 2 (Figure 1) was found to have a heterozygous variant of unknown significance in exon 8 at position 429 in which tyrosine was replaced with histidine. Position 429 is located within the serine-threonine kinase domain and is highly conserved across species, which again may indicate functional significance. Information regarding the presence of the variant in family members is not yet available; however, testing of family members has been recommended. To our knowledge, this variant has not been previously described in the literature.
Genetic testing for non-vascular indications
A small number of patients had genetic testing performed for non-vascular indications. Of particular interest is the coexistence of hereditary multiple exostosis and FMD in a single patient. This patient had intimal fibroplasia involving the right renal and left internal carotid arteries, prior aneurysm of the left internal carotid artery, and a known mutation in the exostosin-1 gene. Hereditary multiple exostosis is a rare condition characterized by the presence of multiple osteochondromas. Vascular complications including vessel impingement, rupture, 43 and pseudoaneurysm 44 have been reported in patients with this disease. Although isolated, this is a novel association between hereditary multiple exostosis, a non-vascular connective tissue disorder, and FMD.
The results of our study demonstrate a low diagnostic yield of genetic testing for known vascular connective tissue disorders in a high-risk cohort of patients with FMD evaluated at a quaternary care center. However, genetic testing in this cohort was not comprehensive in that all patients were not tested for abnormalities in all genes discussed. Owing to the retrospective nature of our study, testing for abnormalities in specific genes was largely at the discretion of the FMD program physician, medical geneticist, and genetic counselor. Data regarding the clinical significance of ACTA2 and SMAD3 mutations is relatively recent and sequence analysis of these genes was not available for all patients at the time of their evaluation. Discrepancy in availability was likely responsible for the lower prevalence of these tests in our patient population.
Furthermore, there are additional vascular connective tissue disorders that might be considered for which no patients were tested in our study. Grange and colleagues reported a familial syndrome of arterial occlusive disease consistent with FMD, hypertension, congenital cardiac defects, bone fragility, brachysyndactyly, and learning disabilities, and proposed that individual components may appear as isolated conditions. 45 Although multiple candidate genes were discussed, no data has been presented to support any one as causative. Arterial tortuosity syndrome (ATS) may also be considered in this population and consists of arterial tortuosity, lengthening, aneurysm, and stenosis as well as ventricular hypertrophy, skin hyperextensibility, and joint laxity.46,47 Coucke and colleagues recently reported that the syndrome was caused by a mutation in the SLC2A10 gene, which encodes the glucose transporter GLUT10. 48 The mutation results in GLUT10 deficiency, and similar to other vascular connective tissue disorders, is associated with enhanced signaling through the TGF-β pathway.
The low yield of genetic testing for Mendelian vascular connective tissue disorders in this study suggests that FMD is a distinct disease entity and not a non-specific finding present in the setting of another well-described vascular connective tissue disease (e.g. EDS type IV). Our results identified two patients with a similar clinical phenotype who had novel and distinct variants in the TGFβR1 gene. These variants are not known to be causative of Loeys-Dietz syndrome, are currently of uncertain clinical significance, and therefore merit further investigation. In addition, our findings highlight the need for additional genetic and molecular research to elucidate the unique genetic and environmental factors associated with the pathogenesis of FMD.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Gornik is a volunteer (non-compensated) medical advisory board member to the Fibromuscular Dysplasia Society of America. All other authors declare no conflicts.