
Abstract
The endothelium plays an integral role in the development and progression of atherosclerosis. Hemodynamic forces, particularly shear stress, have a powerful influence on endothelial phenotype and function; however, there is no clear consensus on how endothelial cells sense shear. Nevertheless, multiple endothelial cell signal transduction pathways are activated when exposed to shear stress in vitro. The type of shear, laminar or oscillatory, impacts which signal transduction pathways are initiated as well as which subsequent genes are up- or down-regulated, thereby influencing endothelial phenotype and function. Recently, human studies have examined the impact of shear stress and different shear patterns at rest and during exercise on endothelial function. Current evidence supports the theory that augmented exercise-induced shear stress contributes to improved endothelial function following acute exercise and exercise training, whereas retrograde shear initiates vascular dysfunction. The purpose of this review is to examine the current theories on how endothelial cells sense shear stress, to provide an overview on shear stress-induced signal transduction pathways and subsequent gene expression, and to review the current literature pertaining to shear stress and shear patterns at rest as well as during exercise in humans and the related effects on endothelial function.
Introduction
Hemodynamic forces, including shear stress (SS), are influential factors that affect endothelial cell (EC) phenotype and function. Increased cardiac output and blood flow during exercise modulate hemodynamic forces against the arterial wall, which include circumferential strain and SS. Furthermore, these hemodynamic forces differ throughout the arterial tree owing to bends and bifurcations. The purpose of this review is to examine how EC sense SS, signal transduction pathways in EC which are activated by SS, and examine the effects of various types of SS that are produced in cell culture on EC phenotype and the resulting factors that affect atherosclerotic etiology. Additionally, research examining SS or shear rate (SR) in humans, with specific emphasis on SR patterns during and after exercise, is reviewed.
How do endothelial cells sense shear stress?
Several theories have been proposed as to how endothelial cells sense SS as a physical force and transmit this information as an intracellular chemical signal. These theories include ion channel activation, caveolae-mediated regulation of Ca2+, G-protein-coupled receptor activation, tyrosine kinase receptor activation, adhesive protein activation, glycocalyx elongation, and bending of primary cilia.1,2
Shear stress sensing – overview and integrative theories
Ion channels
There are several flow-responsive ion channels that participate in sensing SS. The sequence of events begins with activation of inward rectifying K+ channels, followed by activation of outward rectifying Cl− channels. The K+ flux initiates transmembrane hyperpolarization and drives Ca2+ entry into the cell;3–5 whereas non-selective cation channels control the magnitude of Ca2+ influx.6,7 Extracellular Ca2+ passage through the cell membrane follows activation of two SS-dependent ion channels, namely P2X purinoceptors and transient receptor potential channels.8–11 P2X purinoceptors are activated by increases in endogenous intracellular ATP, which has been shown to be SS dose-dependent.12–14 The SS-induced augmentation of intracellular ATP appears to be the result of cell surface ATP synthase bound to caveolae and lipid rafts within the cell surface. 15 The influx of Ca2+ into the EC leads to activation of Ca2+-dependent signaling pathways. Endothelium-derived nitric oxide (NO), which modulates vascular tone, flow-dependent dilation, and vascular remodeling, is generated due to the Ca2+-induced release of caveolae-bound endothelial NO synthase (eNOS). 16 Removal of extracellular Na+, as well as the addition of a voltage-gated Na+ antagonist, attenuated the SS-induced activation of extracellular signal-regulated kinase (ERK). 17 These results suggest that this class of voltage-gated ion channels is involved with sensing SS despite concerns raised over the low membrane potential of EC and the slow changes in membrane polarization, both of which argue against the involvement of SS sensing voltage-gated channels. 18
There are currently three theories on how these channels are activated in response to SS. The first theory is that there is a direct physical interaction between blood flow and the ion channel. The ion channel is ‘pushed’ open due to the drag force that is elicited by blood flow, presumably in a non-specific way related to the magnitude of force generated by SS. In a review by Barakat and colleagues, the authors explore this theory by mathematically modeling the energy due to blood flow drag and concluded that blood flow drag energy is far below the energy required to force open the membrane-bound channel. 2
A second theory on how ion channels sense SS is through ion channel interaction with the cytoskeleton, which appears crucial for SS-induced production of NO. 19 As SS changes the mechanical tension on the cytoskeleton, the ion channel that is associated with the cytoskeleton is activated allowing ion flux through the cell membrane. 20 Indeed, mathematical modeling data suggest that flow may deform sensors and cytoskeletal elements, which may initiate SS-induced intracellular signaling cascades; however, the presumed flow patterns were not representative of flow through the arterial system. 21 EC are exposed to both SS and circumferential stretch in vivo, which complicates the ability to decipher the effects of cytoskeleton deformation on EC ion channel conductance. Although the mechanisms underlying these interactions are not well understood, the potential viability of this theory is supported by observations that shear-induced cellular deformation and cytoskeleton reorientation can activate eNOS and increase NO production.4,22
The third prevailing theory on how ion channels sense SS is through changes in cell membrane fluidity. As blood flow moves across the cell surface the viscosity of the lipid bilayer is altered.23,24 Evidence in support of this theory includes observations that reducing EC membrane fluidity by depleting the cellular membrane of cholesterol leads to ERK activation and abolition of the eNOS response to SS.25,26
Although Na+ channels have not been studied at length, these channels may also be involved in detecting SS. Na+ channels activated by flow have been found in mammalian EC, 27 and Na+ influx through voltage-gated Na+ channels has been observed to inhibit shear induced ERK1/2 activation, 17 indicating that EC shear-related signal transduction may be altered by extracellular Na+.
Summary
The physicochemical mechanisms that underlie the transduction of shear forces into effects on endothelial cell transmembrane channel activation and intracellular signaling responses are incompletely understood. The current best evidence supports participation of membrane cytoskeleton and membrane fluidity in this effect.
Shear stress sensing – cell membrane components
Caveolae
The plasma membrane of endothelial cells is internally lined with small invaginations called caveolae, which may work in conjunction with ion channels and Ca2+ signaling, 28 as well as house ATP synthase. 15 A role for caveolae in sensing SS is suggested by observations that flow-induced Ca2+ responses start at the caveolae and then migrate as a Ca2+ wave across the entire cell membrane. 16 Also, caveolae are well recognized as providing functional compartmentalization of eNOS, and caveolae-bound eNOS is released into the cytoplasm in response to the Ca2+ wave allowing eNOS to become phosphorylated and subsequently produce NO. 16
G-protein-coupled receptors
G-protein-coupled receptors have been implicated in ligand-independent mechanotransduction of SS. SS activates bradykinin B2 G-protein-coupled receptors via a conformational change, possibly due to changes in cell membrane fluidity. 29 Mice lacking B2 G-protein-coupled receptors exhibited a blunted response to blood flow. 30 However, purified G proteins have been found to be activated in response to SS without their associated receptor, 31 suggesting that G proteins sense SS independently.
Tyrosine kinase receptors
SS can activate tyrosine kinase receptors (i.e. VEGFR2, Tie-2) through phosphorylation, independent of their cognate ligands.32–35 A possible mechanism for the activation of VEGFR2 is the SS-induced movement of VEGF2 monomers that initiate dimerization of VEGFR2. 33 The phosphorylation of VEGFR2 could also be caused by the SS-induced ATP release from caveolae and lipid raft-bound ATP synthase. 15 Several signal transduction pathways are initiated through the phosphorylation of tyrosine kinase receptors including ERK, c-Jun N-terminal kinases (JNK), PI3-kinase, and Akt. 1 Activation of PI3-kinase and Akt signal transduction pathways are involved with eNOS phosphorylation and subsequent NO production. 36
Summary of intracellular events
A number of signaling pathways and other intracellular events have been shown to contribute to the responses to shear stress. The known interaction of NOS with caveolae is recognized as an important component of the SS response. Signaling through G-protein-coupled receptors and tyrosine kinase receptors contribute as well, but the relative importance of these mechanisms is not known, and it is not known whether these pathways are sufficient to explain the observed responses.
Shear stress sensing – extracellular components
Cell adhesion molecules
Several cell adhesion molecules have been associated with sensing SS. Integrins have been found to be activated by SS and integrin activation in turn subsequently leads to the activation of the Ras-ERK signal transduction pathway.37,38 The integrins may transmit SS signals to the cytoskeleton.39,40 The Ras-ERK signal transduction pathway is also initiated through the SS-induced phosphorylation of the platelet EC adhesion molecule (PECAM)-1 located in cell junctions. 41 Vascular endothelial cadherins may be integral in the activation of PI3-kinase and Akt pathways through the SS-induced phosphorylation of PECAM-1. These cadherins appear to form a tertiary complex with VEGFR2 and PECAM-1 in response to SS. 42 Within this complex, cadherins act as an adaptor to form signaling complexes. 42
Glycocalyx
The glycocalyx is a network of glycosylated and sialated transmembrane proteins that protrude from the EC surface into the arterial lumen and are coiled under no-flow conditions. As blood flow increases, the glycocalyx, with a net negative charge on the surface layer, 43 becomes uncoiled in the direction of flow causing a conformational change which increases Na+ ion binding sites that initiate signal transduction pathways.44,45 Another postulated role of the glycocalyx in SS mechanotransduction is the transmission of SS through the core of the glycocalyx protein, glypican, to the caveolae, 43 where phosphorylation of eNOS likely occurs through the Src pathway. 46
Primary cilia
Another proposed sensing mechanism involves bending of primary cilia located on the EC surface in response to SS. This bending is thought to allow Ca2+ flux by increasing permeability through ion channels resulting in Ca2+-induced signal transduction. 47 Indeed, polycystin-1 and -2, which are localized on EC cilia, are integrally involved with sensing SS in both mouse and human EC, and the resulting SS mechanotransduction activates Ca2+-dependent signaling cascades.48,49 Primary cilia also appear to disassemble in response to laminar SS. 50 Additionally, when primary cilia are found in vivo, they appear to localize in areas which are prone to atherosclerosis, 51 which indicates they may be involved with sensing low SS.
Summary of extracellular events
It is very likely that more than one of these proposed mechanisms explain how EC sense SS, as increases of intracellular Ca2+ can result from several mechanosensor mechanisms. Interactions among these mechanosensors are thought to coordinate the initiation of SS-induced signal transduction pathways which can modify EC phenotype, gene expression, and EC anti-atherogenic functions. 52 Further in vitro research is warranted to determine how multiple sensors collectively interact to detect SS of various magnitudes and flow patterns and how the resulting interaction influences a pro- or anti-atherogenic environment.
Shear stress – intracellular responses
Shear stress-induced signal transduction in endothelial cells
SS activates a number of signal transduction pathways in EC, which are summarized in Figure 1. Several of these pathways have been previously mentioned; other important components of the response include focal adhesion kinase (FAK), 53 Rho family GTPases, 54 PI3-kinase, 55 mitogen-activated protein kinases (MAPKs),56,57 protein kinase C (PKC), 56 and nuclear factor-κB (NF-κB). 58
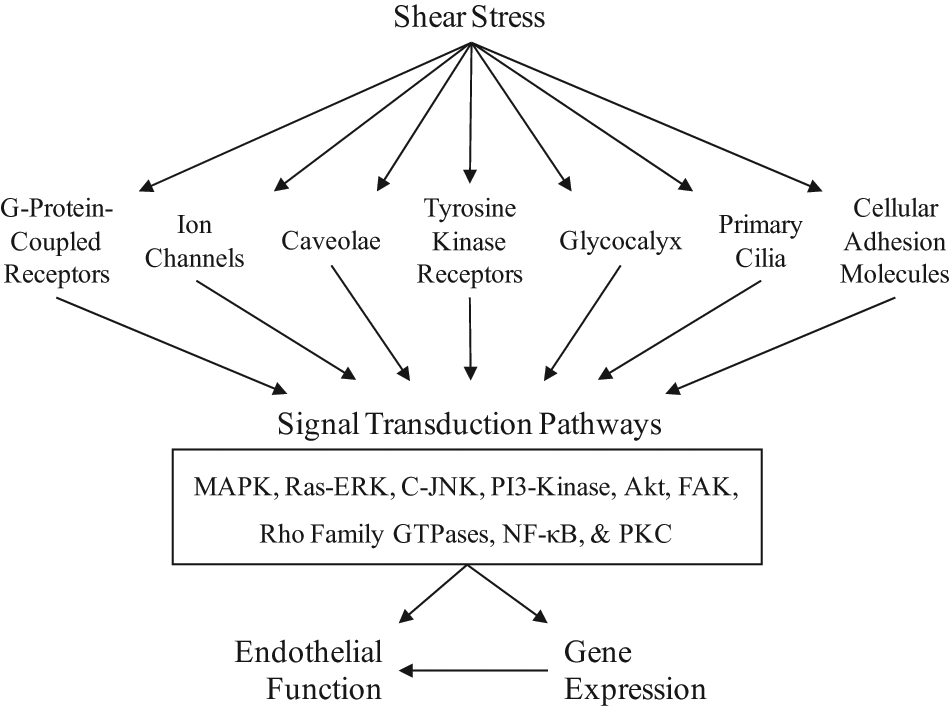
Shear stress appears to be sensed in concert by multiple mechanotransducers located on the membrane of endothelial cells, which initiate several signal transduction pathways that alter gene expression and function. (MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; C-JNK, c-Jun N-terminal kinases; PI3, phosphoinositide 3; FAK, focal adhesion kinase; GTP, guanosine triphosphate; NF-κB, nuclear factor-kappaB; PKC, protein kinase C.)
FAK, a tyrosine kinase, is involved with the activation of the Ras/MAPK pathway and is co-localized with integrins. SS causes an increase in FAK phosphorylation along with FAK activity and it is associated with growth factor receptor binding protein 2/son of sevenless complex, which is involved with Ras/MAPK activation in integrin-mediated cellular adhesion.37,38
SS causes activation of Cdc42 and RhoA, which are members of the Rho family GTPases, which require integrin and extracellular matrix interactions. 59 The major functions of this family of GTPases in response to SS on EC are to activate JNK/activator protein (AP)-1, NF-κB, and c-fos.60–62 c-Fos encodes the AP-1, 63 which appears to regulate endothelin-1 64 and monocyte chemoattractant protein-1 gene expression, 65 and NF-κB controls the expression of inflammatory cytokines, 66 and adhesion molecules. 67
PI3-kinase is rapidly activated by SS in EC, which leads to the production of phosphoinositide 3,4,5- trisphosphate and phosphoinositide 3,4-bisphosphate, which are involved with several downstream signaling pathways. 68 Akt, which is downstream of PI3-kinase, is involved with cellular proliferation, 69 protection against apoptotic stimuli, 70 and eNOS phosphorylation, which leads to NO production in response to SS. 36 Therefore, the PI3-kinase-Akt signaling pathway is vitally important for flow-dependent dilation and providing the anti-atherogenic properties of NO.
MAPKs are a family of Ser/Thr kinases that include p38, ERK and JNK. p38 is involved with MAPK signaling and is activated in response to SS along with ERK 71 and JNK. 72 ERK activation is dependent on kinases and Ca2+ signaling pathways, whereas JNK is mediated through the PI3-kinase/Akt pathway. 55 Activation of the p38 pathway induces the production of pro-inflammatory cytokines 73 and adhesion molecules, 74 and is involved with cellular apoptosis. 75 JNK activation also appears to be involved with cellular apoptosis, and ERK has been implicated in mediating cell growth. 76 MAPKs are involved with several EC gene transcription factors, which include AP-1 57 and c-myc. 77 AP-1 is involved with mediating thrombin activation of endothelin-1 64 and monocyte chemoattractant protein-1 gene expression. 65
The PKC pathway is sensitive to SS in EC. PKC activation by SS subsequently activates ERK downstream 78 along with activating the monocyte chemotactic protein 1 promotor. 79 PKC activation is also involved with gene expression of endothelin-1 80 and platelet-derived growth factor. 81 Endothelin-1 induces vasoconstriction and increases the activity of the renin angiotensin system, sympathetic nerve activity, and macrophage activation, 82 while platelet-derived growth factor has trophic effects on smooth muscle cells and endothelial cells and is implicated in atherogenesis. 83
NF-κB is a transcription factor located in the cytoplasm of EC and is tonically inhibited by binding to the inhibitor of κB (IκB). SS causes the phosphorylation of IκB, which subsequently dissociates from NF-κB. 84 The free NF-κB translocates to the nucleus of the EC to control the expression of cytokines (i.e. IL-6 and IL-8) and adhesion molecules (i.e. ICAM-1 and VCAM-1) involved in the atherosclerotic process. 85
SS alters the expression of approximately 3000 endothelial cell genes. 86 In general, SS induces EC expression of genes that affect growth factors, adhesion molecules, vasoactive substances, endogenous antioxidants, coagulation factors, and chemoattractants. mRNA of growth factors which are increased in response to EC SS are platelet-derived growth factor-A and -B, 87 basic fibroblast growth factor, 88 heparin-binding epidermal growth factor-like growth factor, 89 and transforming growth factor-β. 90 Adhesion molecule gene expression is down-regulated in response to SS. Vascular adhesion molecule-1 (VCAM-1) 91 is down-regulated in response to steady SS, which results in a decrease of leukocyte adhesion to the vascular wall.1,52,67 Vasodilator production of NO92,93 and prostacyclin 94 are increased in response to steady SS and the vasoconstrictor endothelin-1 is decreased. 95 Anti-thrombotic genes are also up-regulated after EC exposure to steady SS. Tissue plasminogen activator,96,97 thrombomodulin, 98 and cylcooxygenase-2 99 gene expression are increased in response to sustained SS. Two forms of superoxide dismutase (SOD) (Mn and Cu/Zn) genes are increased following SS exposure, which enhances the capacity to mitigate reactive oxygen species.99–103–103 In summary, SS tends to induce EC gene expression, which results in an anti-atherogenic environment. However, the expression of some of these EC genes can be differentially regulated when patterns of SS are manipulated or when blood flow is disturbed at bifurcations in the arterial tree.
Methods of applying shear stress in cell culture
Cone and plate flow systems
Manipulating SS patterns in cell culture can be performed by using a cone and plate flow system. This system utilizes a Teflon cone positioned in the center of a tissue culture dish. 104 The tip of the cone is placed in the medium and the cone is rotated to produce uniform flow conditions across the cultured endothelium. The SS is calculated as follows:
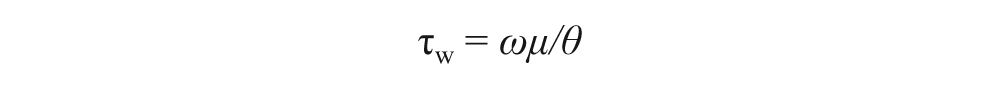
where ω is the rotation speed, µ is the viscosity of the fluid, and θ is the angle of the cone. 104
Parallel plate flow systems
The most common method of subjecting SS to cultured cells is the parallel plate flow system. 104 A gasket is placed on the bottom of a tissue culture dish with a standard rectangular cut-out channel which can be modified to induce various areas of disturbed flow within the flow domain. Inlet and outlet ports are located at either end of the rectangular cut-out flow domain in order to create flow across the cultured EC. The SS applied to the endothelium is calculated as follows:
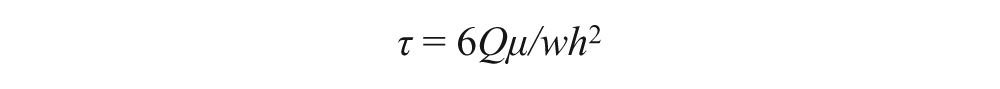
where Q is flow, µ is the fluid viscosity, w is the width of the channel, and h is the thickness of the gasket. 104
Types of shear stress
Endothelial cells are subjected to various forms and magnitudes of SS in vivo. Mimicking these patterns produces differences in the resulting EC phenotypes and in the factors that are produced by EC due to SS, differing by the type of SS applied. Laminar blood flow produces predominantly antegrade SS along the EC surface, whereas bifurcations in the arterial tree disturb the laminar blood flow to produce areas of low SS, retrograde SS, and oscillatory SS.
Laminar blood flow
Laminar blood flow is characteristic of steady, undisturbed blood flow that creates a constant SS along the EC surface. A steady laminar flow activates both K+ and Cl− channels that leads to depolarization of the cell membrane. 105 Flow-dependent K+ channels are initially activated and reach peak activation at different magnitudes of SS than Cl− dependent channels. 105 EC may sense the magnitude of SS based on the relative amplitudes of the K+ and Cl− currents 105 or through the SS-induced dose-dependent influx of Ca2+ through cation channels.12,13,106 The influx of Ca2+ into the EC causes activation of PKC and subsequent activation of MAPKs, which modulate transcription factors and gene expression along with other signal transduction pathways which may be activated through additional methods of SS detection. 52
EC exposed to laminar blood flow and SS typically exhibit an anti-atherogenic phenotype that is accompanied by expression of anti-atherogenic genes. One of the most important anti-atherogenic molecules produced in vivo is NO, which is catalyzed by eNOS in response to SS, insulin, and acetylcholine. eNOS mRNA expression in cultured EC is up-regulated in response to 1 hour of laminar SS. 107 Isolated and cannulated porcine coronary arterioles were subjected to three doses of laminar SS (no shear, low shear, and high shear) for 2 hours and 4 hours, and only high SS induced an increase of eNOS mRNA following both time periods. 108 Additionally, exercise training has been shown to increase eNOS protein and activation through a SS-dependent mechanism;100,109,110 however, the increased SS during and after a single session of exercise has antegrade and retrograde flow patterns that create oscillatory shear across the EC surface. 111 The increased production of NO through the up-regulation and activity of eNOS produces an anti-atherogenic environment as NO inhibits several aspects of atherogenesis, including smooth muscle cell proliferation and migration, platelet aggregation and leukocyte adhesion to the vascular wall, improves fibrinolysis, regulates permeability and vasomotor tone, as well as act as an antioxidant under situations of increased superoxide anion concentrations. 112
As previously mentioned, SS increases SOD mRNA and protein content to reduce oxidative stress, a key component in atherogenesis. Indeed, Woodman and colleagues observed an increased SOD mRNA in porcine arterioles following 4 hours of high laminar SS; whereas stagnant SS and low SS conditions did not affect SOD mRNA. 108 However, the increased antioxidant capacity may be a secondary response to increased superoxide radical production by SS-stimulated nicotinamide adenine dinucleotide (NADH) oxidase, 113 and not necessarily a direct effect of SS on EC.
Atherosclerosis progression is also mediated through the coagulation cascade, which is also affected by laminar SS. As previously mentioned, tissue plasminogen activator, thrombomodulin, and cylcooxygenase-2 gene expression are up-regulated and plasminogen activator inhibitor type-1 is down-regulated in response to steady SS. SS also rapidly induces the production of the vasodilator NO, which appears to inhibit platelet aggregation by increasing platelet cyclic GMP and through the S-nitrosylation of platelet proteins. 114
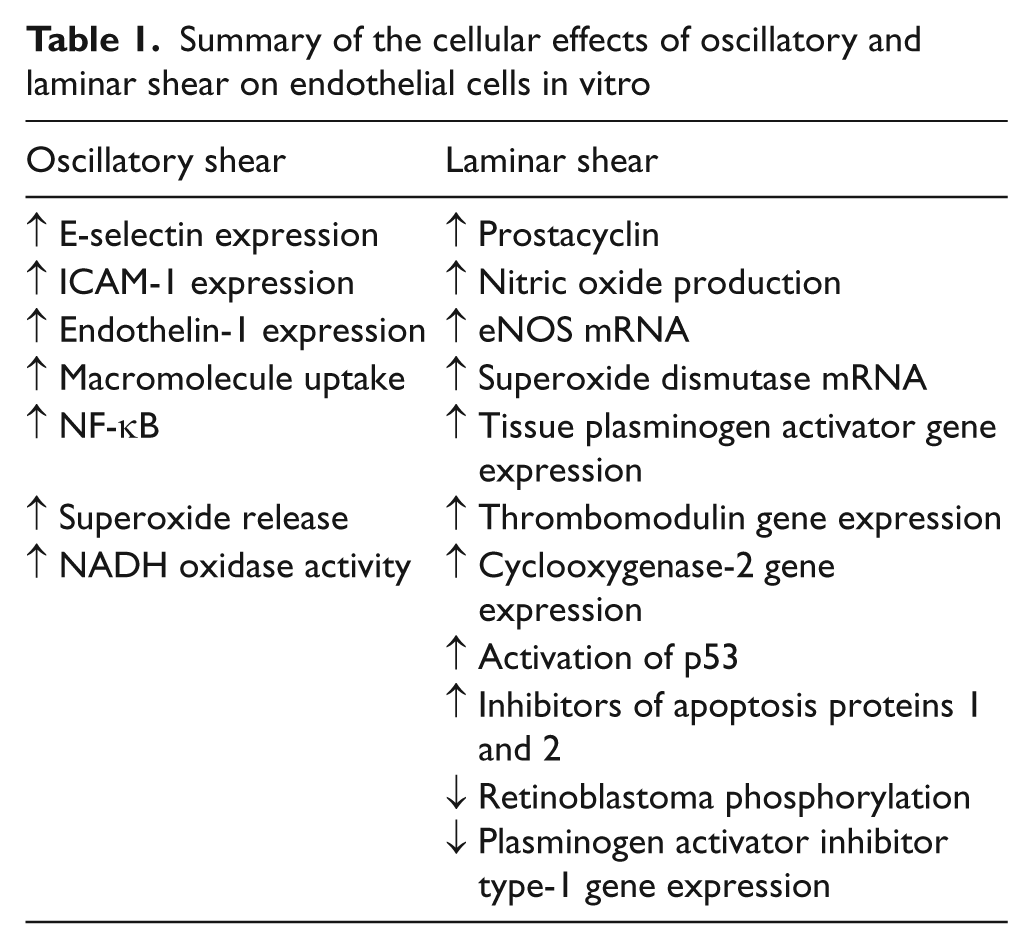
Laminar SS has profound effects on EC proliferation and apoptosis. Twenty-four hours of laminar SS has been observed to be a potential inhibitor of EC proliferation by activating p53, which increases growth arrest proteins and reduces EC proliferation. 115 Exposed to 18 hours of SS-suppressed EC apoptosis despite being treated with pro-apoptotic stimuli such as TNF-α, oxygen radicals, and oxidized LDL, through inhibition of interleukin 1β-converting enzyme and capase-3. 116 The PI3-kinase/Akt survival pathway has also been observed to be activated following 1 h of SS exposure. 70 Furthermore, the up-regulated activity of Cu/Zn SOD and eNOS due to SS appears to suppress cellular apoptosis. 100 Inhibitor of apoptosis protein 1 is up-regulated in response to 24 hours of laminar SS, 117 whereas inhibitor of apoptosis protein 2 is increased following 4 hours of laminar SS. 118 Retinoblastoma protein, a cellular regulator of cell cycles, is hypophosphorylated in response to 24 hours of laminar SS, thus reducing cellular proliferation. 115 These effects collectively produce an anti-atherogenic environment by reducing EC turnover, thus preventing lesions in straight segments of the arterial tree where laminar flow is prevalent. A summary of the cellular effects of laminar SS on endothelial cells in vitro is provided in Table 1.
Summary of the cellular effects of oscillatory and laminar shear on endothelial cells in vitro
Oscillatory blood flow
Bifurcations in the arterial tree modify laminar blood flow and SS into areas of low SS and oscillatory flow patterns beyond the bifurcation. These areas of low and oscillatory SS are prone to atherosclerotic lesions. 119 Cheng and colleagues manipulated the SS pattern in vivo. 120 Mice were outfitted with a cast on the carotid artery to manipulate SS to produce areas of high laminar SS, low SS and oscillatory SS while on an atherogenic diet. The areas of low SS produced vulnerable atherosclerotic lesions whereas oscillatory SS produced stable atherosclerotic lesions after 6 weeks of the atherogenic diet. 120 The low SS areas also had larger lesions which contained more lipids, fewer smooth muscle cells, less collagen, and protruded less into the lumen, produced more pro-inflammatory mediators and intraplaque hemorrhages when compared to the oscillatory regions. On the other hand, the areas of high laminar SS did not exhibit atherosclerotic lesions following 6 weeks of the experiment. 120 This SS manipulation in vivo provides potent evidence for the negative effects of oscillatory and low SS.
The frequency of oscillatory SS may have important implications in EC function and gene expression. Oscillatory SS with frequencies of 0.2 or 1.0 Hz have been shown to fully activate K+ channels while only minimally affecting Cl− channels and hyperpolarizing the cell membrane. 105 A greater frequency of oscillatory SS, 5 Hz has been shown to not activate either K+ or Cl− channels. A high frequency of oscillatory SS with little net directional flow may be too fast for the EC ion channels to detect 2 as there is no net influx of Ca2+ into the cell during periods of pure oscillation. 121 The recognition of oscillatory SS by EC may be sensed by the hyperpolarized cell membrane which in turn induces the pro-atherogenic environment.
Oscillatory flow has been shown to induce the expression of ICAM-1, E-selectin, and endothelin-1, increase monocyte-EC attachment through NF-κB, as well as increase EC macromolecule uptake; whereas steady laminar flow down-regulates VCAM-1 and endothelin-1. 52 The expression of ICAM-1 and VCAM-1 may be sensitive to the oxidative state of the cell as antioxidant treatment of N-acetylcysteine prevented the 24-hour oscillatory SS-induced expression of VCAM-1 and reduced ICAM-1 expression by nearly 70%. 122 Indeed, oxidative stress is induced by oscillatory SS through the increase of intracellular superoxide radicals and NADH oxidase activity. 113 It has also been suggested that NAD(P)H oxidase maintains the function of xanthine oxidase during periods of oscillatory SS to produce superoxide radicals in EC. 123 The complex interaction between oxidative stress and adhesion molecule gene expression during periods of oscillatory SS underscore the importance of reducing superoxide radicals in EC by increasing antioxidant capacity or reducing superoxide generation. A summary of the cellular effects of oscillatory SS on endothelial cells in vitro is provided in Table 1.
The recognition and transmission of SS to biochemical processes in EC is a complex and integrated series of events which generates a wide range of pro- and anti-atherogenic functions and phenotypes contingent upon the type of SS exposure. Additional research that examines the regulation and interaction of downstream intracellular signaling pathways which affect EC phenotype and function in response to SS which accurately mimics in vivo patterns merits further investigation.
Although extensive research has focused on SS in vitro, the extrapolation of these results to in vivo events has its limitations. In vitro research commonly attempts to mimic the vascular milieu in vivo; however, it may over simplify the effects of SS on EC as the combined magnitude of SS and the corresponding circumferential strain, or lack thereof, may not be applicable in vivo. Furthermore, in vitro research does not account for additional mechanisms which may regulate EC phenotype and function in vivo. Estimated SS at rest in mice conduit arteries (1.2–8.4 Pa) 124 is far greater than shear exhibited in human conduit arteries (0.4–1.3 Pa), 125 further confounding the extension of animal results to humans.
Human studies
The data presented above were derived from studies conducted on EC from various animals or human umbilical veins in cell culture. We now turn to studies on SS and SS patterns at rest as well as during and following aerobic exercise in humans and, where applicable, how the SS affects endothelial function. However, SS is not commonly assessed in humans due to the difficulty of measuring the viscosity of blood in direct contact with the endothelium. Owing to this restraint, shear rate (SR) is used as an estimation of SS in humans and is generally defined as blood velocity divided by arterial diameter. Arterial blood velocity (both antegrade and retrograde) and diameters are typically viewed using a duplex ultrasound which images simultaneous pulsed-wave Doppler blood velocities and two-dimensional B-mode diameters. 111 Additional Doppler envelope detection and arterial wall tracking software is used to analyze the magnitude of antegrade and retrograde blood velocities and measure arterial diameters, respectively.
Shear rate in conduit arteries feeding the working limbs
Tinken and colleagues performed a study to address the significance of exercise-induced SR in the brachial artery following 8 weeks of bilateral handgrip exercise for the improvement of flow-mediated dilation (FMD), 126 which is an assessment of endothelium derived NO127–130 (Table 2). One forearm served as the non-cuffed control, whereas the contralateral arm had a blood pressure cuff inflated to 60 mmHg to alter the magnitude of exercise-induced SR. Mean SR, antegrade SR, and retrograde SR were increased above baseline in the non-cuffed arm whereas the cuffed arm did not receive a change in mean, antegrade, or retrograde SR during acute handgrip exercise. 126 An increased FMD was seen in the non-cuffed arm at weeks 2, 4, and 6. The cuffed arm maintained the same FMD throughout the 8-week training program. 126 These findings emphasize the importance of augmented SR during exercise as a means to improve FMD. Studies which have focused on the effects of shear on FMD are summarized in Table 2.
Summary of the effects of shear in humans on endothelial function
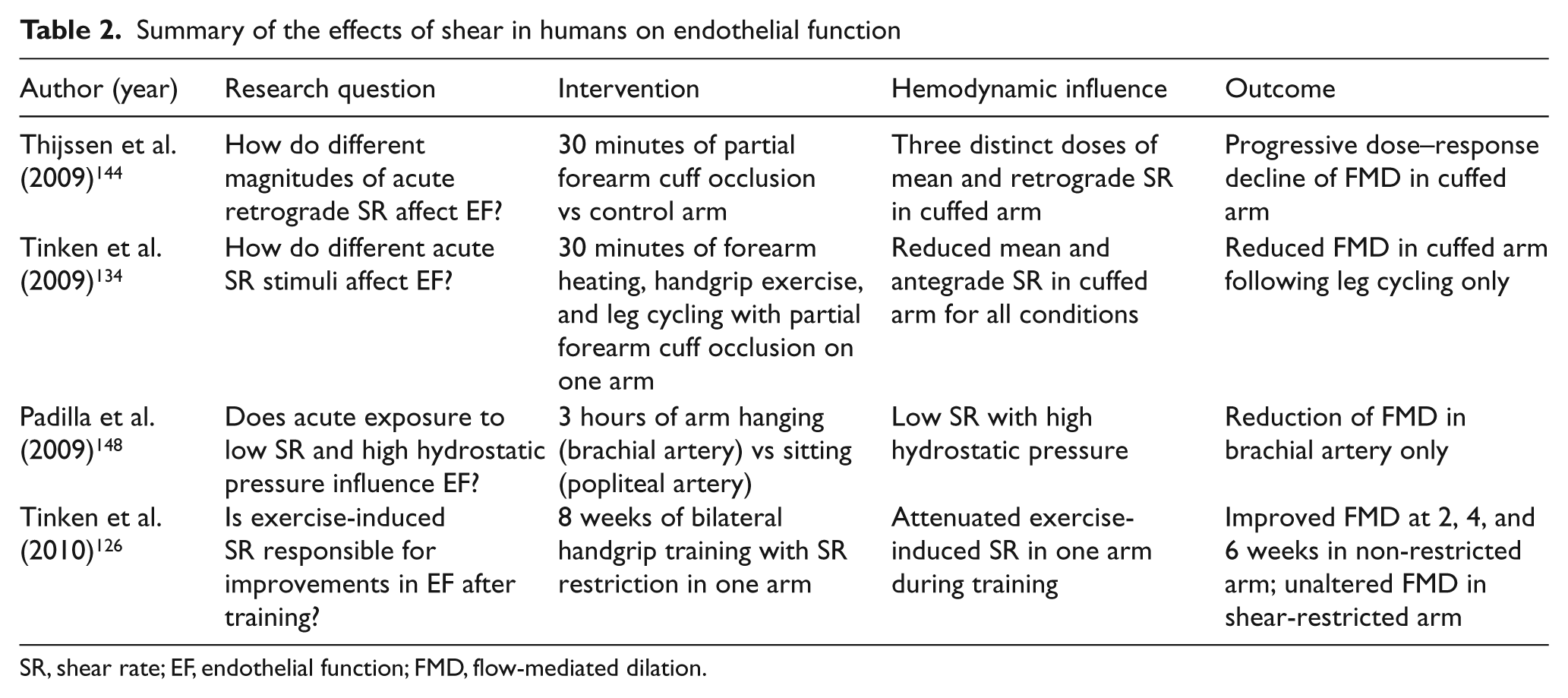
SR, shear rate; EF, endothelial function; FMD, flow-mediated dilation.
Shear rate in conduit arteries feeding non-working limbs during exercise
Blood flow and SR increase in the non-exercising limbs during exercise, but not nearly to the same extent as in exercising limbs.111,131–133 It is postulated that this increased SR to non-exercising limbs during exercise is partially responsible for the improvement in vascular function assessed in non-exercise trained limbs.134,135
The magnitude of SR in non-working limbs during exercise appears to be dependent on exercise intensity. Antegrade blood flow through the brachial artery was measured during incremental lower body cycle exercise and significantly increased at a somewhat low power output of 60 W and continued to increase up to 160 W (end of data collection). 111 Green and colleagues had also found that brachial artery retrograde blood flow progressively increased, starting at 40 W during 3-minute stages of incremental cycle ergometer exercise. 111 Tanaka and colleagues found a similar increase in brachial artery blood flow and SS starting at 60 W during 3-minute stages of incremental lower body cycle ergometer exercise, and found a progressive increase in femoral blood flow and SS starting at 20 W during 5-minute stages of incremental arm crank exercise. 133 Five 3-minute stages were used to observe dose-dependent increases of brachial artery mean blood flow and SR during walking at low speeds (3, 4, and 5 km/h), repeated kicking with three different loads (5 kg, 7.5 kg, and 10 kg), and cycling at three various workloads (60 W, 80 W, and 120 W). 131 Three 30-minute interventions (forearm heating, handgrip exercise, and lower body cycling) with partial lower arm occlusion randomly placed on one arm were performed by healthy volunteers 134 (Table 2). Although retrograde flow was not significantly altered in the two exercise treatments, mean SR and antegrade SR were both significantly reduced due to the lower arm occlusion. The change in SR patterns during the exercise treatments altered local FMD. The non-cuffed arm significantly increased FMD following both exercise conditions, whereas the cuffed arm during cycling exercise had decreased FMD and the cuffed arm during handgrip exercise was not significantly altered. 134 These results suggest that mean and antegrade SR increase in an exercise intensity dose-dependent manner during exercise of short duration (3–5 minutes), and reduced mean or antegrade SR during exercise can directly impact endothelial function. These results also underscore the importance of increased SR during exercise for improved FMD.
Exercise intensity is associated with the magnitude of antegrade SR in non-working limbs and thus high-intensity exercise may generate SR, which produces the greatest improvement in endothelial function. However, this is not necessarily the case. Acute high-intensity exercise has been observed to increase oxidative stress 136 and exercise training has been shown to elicit decreased circulating antioxidants. 137 A decreased endothelial function following both acute and chronic high-intensity exercise have also been reported.136,137 Conversely, acute moderate intensity exercise appears to reduce oxidative stress and increase FMD, 136 and moderate intensity exercise training appears to augment antioxidant defense 138 and endothelial function. 139 Collectively these results suggest that the hormesis theory for adaptation applies to antioxidant and endothelial alterations following acute and chronic exercise.140,141
The increased oxidative stress from high-intensity exercise bouts may be initiated by the augmented oscillatory and retrograde SS that is associated with greater work rates. Oscillatory SS has been observed to increase superoxide generation in cultured EC by increasing NADH oxidase and xanthine oxidase activity.113,123 Retrograde blood flow and SR have been reported to be exercise intensity-dependent to the same extent as antegrade blood flow. Green and colleague’s earlier work observed the progressive increase in retrograde blood flow and SR in the brachial artery during incremental cycle exercise, which started slightly earlier in the protocol (40 W) when compared to antegrade blood flow (60 W).111,132 Walking is also a mode of exercise that progressively increases brachial artery retrograde blood flow and SR with increasing exercise intensity, which elicits a greater oscillatory SS on the EC surface. 131 However, retrograde SS in the brachial artery during leg kicking does not appear to increase with exercise intensity. 131 This mode of exercise provides a possible model to examine the effect of exercise training, without the impact of oscillatory SR, on endothelial function. It could be speculated that if indeed repeated bouts of oscillatory SR negatively affect long-term EC function, then leg kick training may exhibit greater EC function improvements when compared to other modes of training.
The effects of oscillatory shear at rest on endothelial function
Oscillatory SR has been identified as a possible mechanism for reduced endothelial function. Healthy older adults tend to exhibit greater retrograde and oscillatory SR in the femoral artery 142 and typically have lower femoral artery endothelial function when compared to healthy young adults. 143 This suggests that increased retrograde blood flow in older adults is part of the natural aging process and that the augmented retrograde blood flow may have a negative impact on endothelial function. Thijssen et al. manipulated blood flow by partial forearm occlusion to induce three distinct doses of increased retrograde blood flow and SR in healthy men at rest 144 (Table 2). There was a progressive decrease of FMD that corresponded to the increased retrograde SR. A significant correlation between the change of FMD and the change of retrograde SR was also found. 144 These results indicate that increased retrograde SR at rest, which increases oscillatory SR, causes a relative reduction of FMD. Table 2 summarizes the effects of oscillatory SR on endothelial function in humans.
Potential modulators of shear rate profiles
What modulates retrograde and oscillatory SR? Thijssen et al. recently demonstrated that healthy, able-bodied controls and spinal cord-injured subjects display similar SR patterns in the femoral artery during arm crank exercise, thus possibly ruling out increased sympathetic nervous system activation to the non-working limbs. 145 However, Padilla and colleagues subjected healthy men to several sympathoexcitatory maneuvers which resulted in augmented muscle sympathetic nerve activity, retrograde and oscillatory shear patterns, as well as an increase in arterial blood pressure. 146 Recent evidence also suggests that thermoregulatory vasodilation of the forearm vasculature modulates the magnitude of retrograde SR during prolonged leg cycling. 147 This leads to speculation that results obtained by Green et al., 111 Tanaka et al., 133 and Thijssen et al. 131 cannot be extended beyond the 3–5-minute data collection periods. Collectively, these findings indicate that there may be an interaction among the degree or duration of sympathetic activity (or lack thereof), arterial blood pressure, and thermoregulatory mechanisms which may alter shear patterns. Alternative theories include increased blood flow reflection due to amplified systolic arterial pressure accompanied by augmented antegrade velocity, or an enhanced myogenic response during diastole due to heightened systolic arterial pressure during exercise. 145
Shear rate and hydrostatic pressure
A reduction of shear with an accompanying increased hydrostatic pressure also impacts endothelial function at rest; however, the response appears to be limb-specific. Padilla et al. investigated the impact of acute arm hanging (a low SR combined with high arterial pressure used to mimic the vascular conditions of the lower leg in a seated position) in the brachial artery and acute sitting in the popliteal artery on FMD 148 (Table 2). The observed response was limb-specific; the brachial artery exhibited a reduction of FMD, whereas FMD in the popliteal artery remained unchanged. 148 These findings suggest a possible adaptation in the popliteal artery to these vascular conditions and a possible increased sensitivity to the hemodynamic conditions observed in the brachial artery. 148
Role of nitric oxide production during dynamic aerobic exercise
NO appears to be vital in allowing SR to increase in the brachial artery during lower body exercise. Green et al. infused the eNOS inhibitor
Brachial artery shear rate following aerobic exercise
The increased SR during exercise appears to play a major role in the prevention of atherosclerosis; furthermore, SR appears to remain elevated following acute bouts of aerobic exercise as well. Despite the possible beneficial role of elevated post-exercise SR in endothelial function, literature is presently scarce. Post-exercise SR was characterized by Padilla et al. following 45 minutes of low, moderate, and high-intensity exercise in older, overweight or obese men. 150 High-intensity exercise produced the greatest SR when compared to low and moderate intensities. Unfortunately, baseline SR and retrograde SR were not reported and thus it is unclear if and/or when SR returned to pre-exercise values within the 3-hour window and if post-exercise retrograde SR was altered. 150
Endothelial function in conduit arteries which feed both the working and non-working limbs following acute exercise and exercise training appears to be heavily influenced by the magnitude and pattern of the exercise-induced SR. Additional research is warranted to investigate the relationship between SR magnitude and patterns during various exercise modalities and the degree to which the exercise-induced SR exerts anti- or pro-atherogenic functions of the endothelium. Furthermore, potential in vivo mechanisms (i.e. oxidative stress, adhesion molecule expression, cytokines) responsible for altering endothelial function following periods of augmented retrograde SR deserve attention to further validate in vitro findings.
Summary
Endothelial cells appear to sense shear stress through several different mechanisms, which modulate many signal transduction pathways to influence cell phenotype and function. The type of shear stress to which the endothelial cell is exposed determines the shear-induced changes in intracellular signaling and gene expression, which in turn elicits a spectrum of pro- or anti-atherogenic effects on the endothelial cell. Arteries exposed to retrograde and oscillatory shear stress appear to activate the endothelium in vitro; these types of shear patterns appear to impair endothelial function in humans. Further studies are needed to investigate approaches to modulating shear stress with the goals of mitigating adverse effects of unfavorable forms of shear and improving vasodilator and anti-atherosclerotic functions of the vascular endothelium.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.