
Abstract
Background:
Amiselimod, an oral selective sphingosine-1-phosphate 1 receptor modulator, suppressed disease activity dose-dependently without clinically relevant bradyarrhythmia in a 24-week phase 2, placebo-controlled study in relapsing-remitting multiple sclerosis.
Objective:
To assess safety and efficacy of amiselimod over 96 weeks.
Methods:
After completing the core study, patients on amiselimod continued at the same dose, whereas those on placebo were randomised 1:1:1 to amiselimod 0.1, 0.2 or 0.4 mg for another 72 weeks. Most patients receiving 0.1 mg were re-randomised to 0.2 or 0.4 mg upon availability of the core study results.
Results:
Of 415 patients randomised in the core study, 367 (88.4%) entered and 322 (77.6%) completed the extension. One or more adverse events were reported in 303 (82.6%) of 367 patients: ‘headache’, ‘lymphocyte count decreased’, ‘nasopharyngitis’ and ‘MS relapse’ were most common (14.7%–16.9%). No serious opportunistic infection, macular oedema or malignancy was reported and no bradyarrhythmia of clinical concern was observed by Holter or 12-lead electrocardiogram. The dose-dependent effect of amiselimod on clinical and magnetic resonance imaging-related outcomes from the core study was sustained in those continuing on amiselimod and similarly observed after switching to active drug.
Conclusion:
For up to 2 years of treatment, amiselimod was well tolerated and dose-dependently effective in controlling disease activity.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic neurological disease that requires long-term treatment. 1 Therefore, safety and efficacy of potential new treatments need to be assessed over long periods of time.
Amiselimod (also known as MT-1303) is an oral selective sphingosine-1-phosphate (S1P) 1 receptor modulator currently being developed for the treatment of various autoimmune diseases. 2 A 24-week phase 2, randomised, placebo-controlled dose-ranging study (MOMENTUM; ClinicalTrials.gov, NCT01742052) demonstrated safety and dose-dependent efficacy of once-daily oral amiselimod in patients with relapsing-remitting multiple sclerosis (RRMS). 3
Here, we report the results of MOMENTUM Extension (hereafter referred to as ‘Extension’), a 72-week dose-blinded safety and efficacy extension study to the MOMENTUM phase 2 (hereafter referred to as ‘Core’) study in patients with RRMS. This extension study (ClinicalTrials.gov, NCT01890655) aimed to evaluate the long-term safety and tolerability and also the long-term effects of amiselimod on pharmacodynamics (PD), magnetic resonance imaging (MRI)-related and clinical outcomes including health-related quality of life (QoL).
Methods
Study design
After completion of the 24-week treatment in the Core study, patients who provided written informed consent entered the 72-week Extension study (Figure 1(a)). Patients on amiselimod in the Core study continued at the same dose in the Extension double-blind period (Part 1). Those originally on placebo were equally randomised to amiselimod 0.1, 0.2 or 0.4 mg. Based on the results of the Core study, all patients on 0.1-mg dose who were ongoing in the Extension Part 1 with a minimum of 12 weeks left in the Extension study were equally re-randomised to either amiselimod 0.2 or 0.4 mg on entry to the Extension open-label period (Part 2). After completion of Part 2, patients entered the 12-week Safety Follow-up period.
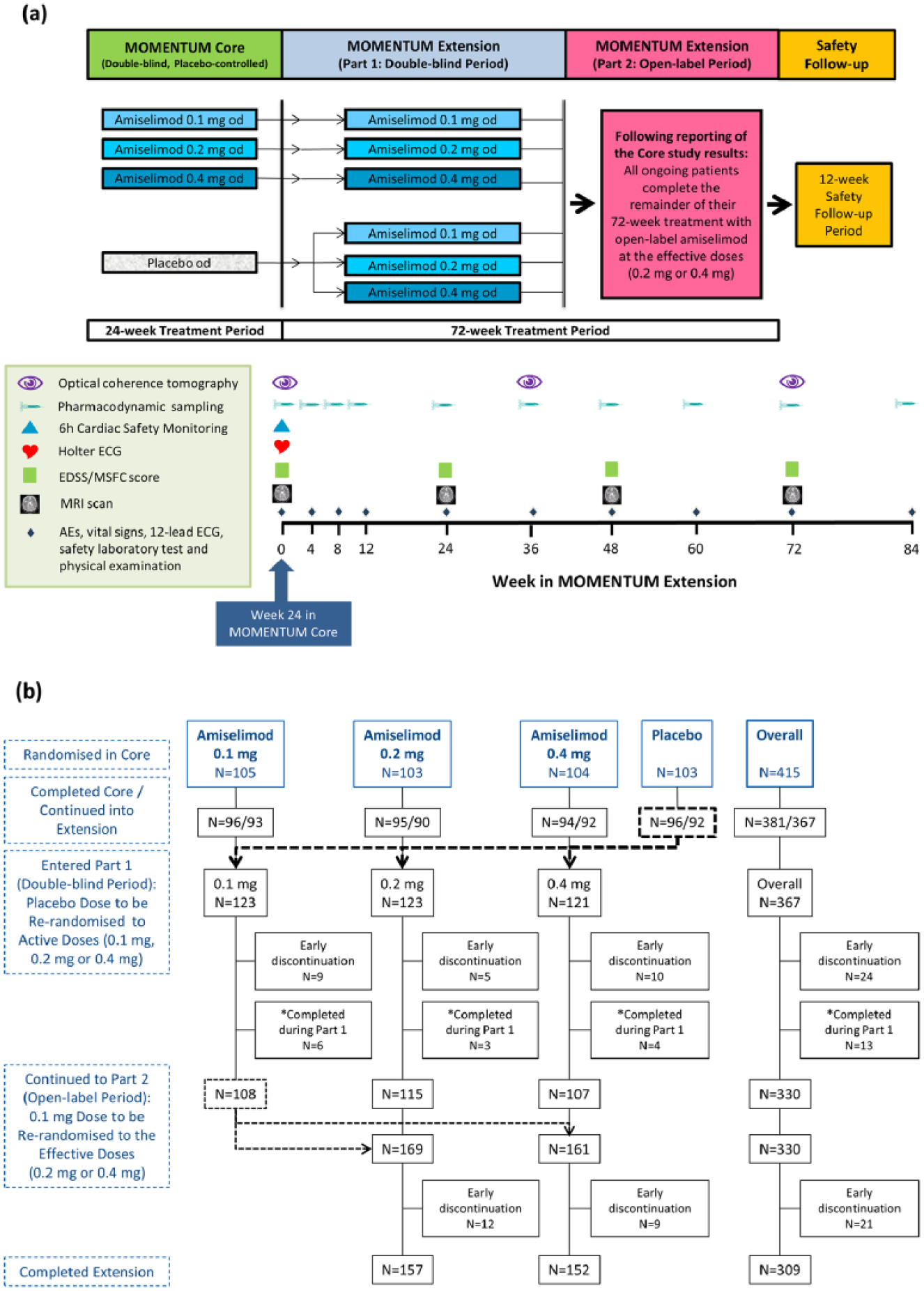
MOMENTUM study design and patient disposition. (a)
This study was approved by the respective independent ethics committees of all participating sites and conducted in accordance with the Declaration of Helsinki, International Conference of Harmonisation and Good Clinical Practice guidelines, local regulations and the European Medicines Agency Guideline 4 (a list of the principal investigators is provided in ‘MOMENTUM Study Group’).
Study objectives
The primary objective of the Extension study was to evaluate the long-term safety and tolerability of amiselimod in patients with RRMS. Secondary objectives were to assess the long-term effects of amiselimod on PD, MRI-related and clinical outcomes including health-related QoL.
Study procedures and assessments
In the Extension study, patients attended the study sites every 4 weeks until Week 12 and every 12 weeks thereafter until the Safety Follow-up visit at Week 84 (Figure 1(a)). Safety and tolerability of amiselimod was assessed by monitoring adverse events (AEs), vital signs (blood pressure and pulse rate), 12-lead electrocardiogram (ECG), safety laboratory tests (haematology, biochemistry and urinalysis) and physical examination at every visit. Optical coherence tomography (OCT) was performed every 36 weeks. On the first dosing day in the Extension study (Week 0), all patients were required to stay in the site for at least 6 hours with hourly assessment of vital signs and 12-lead ECG. In addition, 3-lead Holter ECG was performed for 24 hours. As previously reported,3,5 mean hourly heart rate (HR) was used to assess a potential negative chronotropic effect, whereas pre-defined arrhythmia criteria were used to evaluate a potential negative dromotropic effect. All 12-lead and Holter ECG data were centrally analysed and reported by independent, treatment-blind board-certified cardiologists at Quintiles Cardiac Safety Services (Mumbai, India).
The annualised relapse rate (ARR), time to first confirmed relapse and proportion of patients who remained relapse-free by the end of treatment (Week 72) were evaluated. The level of impairment or disability was assessed using the Expanded Disability Status Scale (Neurostatus-EDSS)6,7 and the Multiple Sclerosis Functional Composite (MSFC) 8 score every 24 weeks by specially trained and certified 7 examiners who were not otherwise involved in the care of the patients. These examiners were also responsible for confirmation of protocol-defined MS relapses at unscheduled visits. Patients’ health-related QoL was assessed with the Multiple Sclerosis Quality of Life-54 (MSQOL-54) score 9 at Week 72. Brain MRI scans were performed every 24 weeks until Week 72. MRI data were centrally analysed and reported by independent, treatment-blind experts at NeuroRx Research (Montreal, QC, Canada). 3 The PD effect of amiselimod was evaluated by measuring peripheral lymphocyte counts at every visit.
The independent Data Safety Monitoring Board (DSMB) appointed prior to the start of the Core study 3 continued to act as DSMB for the Extension study (a list of DSMB members is provided in ‘MOMENTUM Data Safety Monitoring Board’). The DSMB reviewed safety data at pre-defined intervals and assessed the overall benefit–risk profile during the study. In addition, Holter ECGs were independently reviewed during the study by the DSMB cardiologist (E.A.).
Statistical analysis
Three different types of analysis were performed according to treatment sequence and duration:
The ‘Safety population’ analysis set includes all patients who received at least one dose of study medication. Due to multiple randomisations and variations in treatment and duration, subjects in each group may not have been mutually exclusive and may have had different treatment durations. For these reasons, only limited analyses were done in this set.
The ‘By Treatment Sequence’ analysis set includes patients who received either 0.2 or 0.4 mg of amiselimod throughout Core and Extension (‘continuous treatment groups’) and those who were on placebo treatment in the Core study and received either 0.2 or 0.4 mg of amiselimod in the Extension study (‘placebo-switched groups’). The comprehensive analyses of safety and clinical efficacy were done in this set.
The ‘Duration of Exposure’ analysis set includes patients whose amiselimod dose (either 0.1, 0.2 or 0.4 mg) was not changed for up to 48 weeks of treatment. The efficacy analyses related to MRI outcomes (gadolinium (Gd)-enhanced T1-weighted lesions, new or enlarged T2-weighted lesions, total brain volume and grey matter volume) and the long-term cardiac safety assessment were primarily done in this set.
All analyses included the data obtained during the Core study, and ‘baseline’ was defined as the baseline data in the Core study, that is, the last observed value of the parameter of interest prior to the first administration of study medication (Figure 1(a)). The ARR was analysed using the negative binomial regression model. 3 Time to first confirmed relapse as well as time to 12-week confirmed disability progression was analysed using Kaplan–Meier estimates. All other clinical, MRI-related and safety data were summarised in a descriptive manner. All statistical analyses were conducted using SAS (version 9.2).
Results
Of the 415 patients randomised in the Core study, 381 (91.8%) completed the 24-week treatment period, 367 (88.4%) continued into the Extension study and 322 (77.6%) completed 96 weeks on study medication throughout the Core and Extension studies (Figure 1(b)). Demographics and baseline characteristics of patients entering the Extension study were generally similar to those in the randomised population in the Core study 3 (Table 1). The reasons for early discontinuation of study medication varied, but ‘AEs’ (19 (5.2%) patients overall) were the most common (Table 2).
Demographics and baseline characteristics of patients entering the Extension study – Safety population.
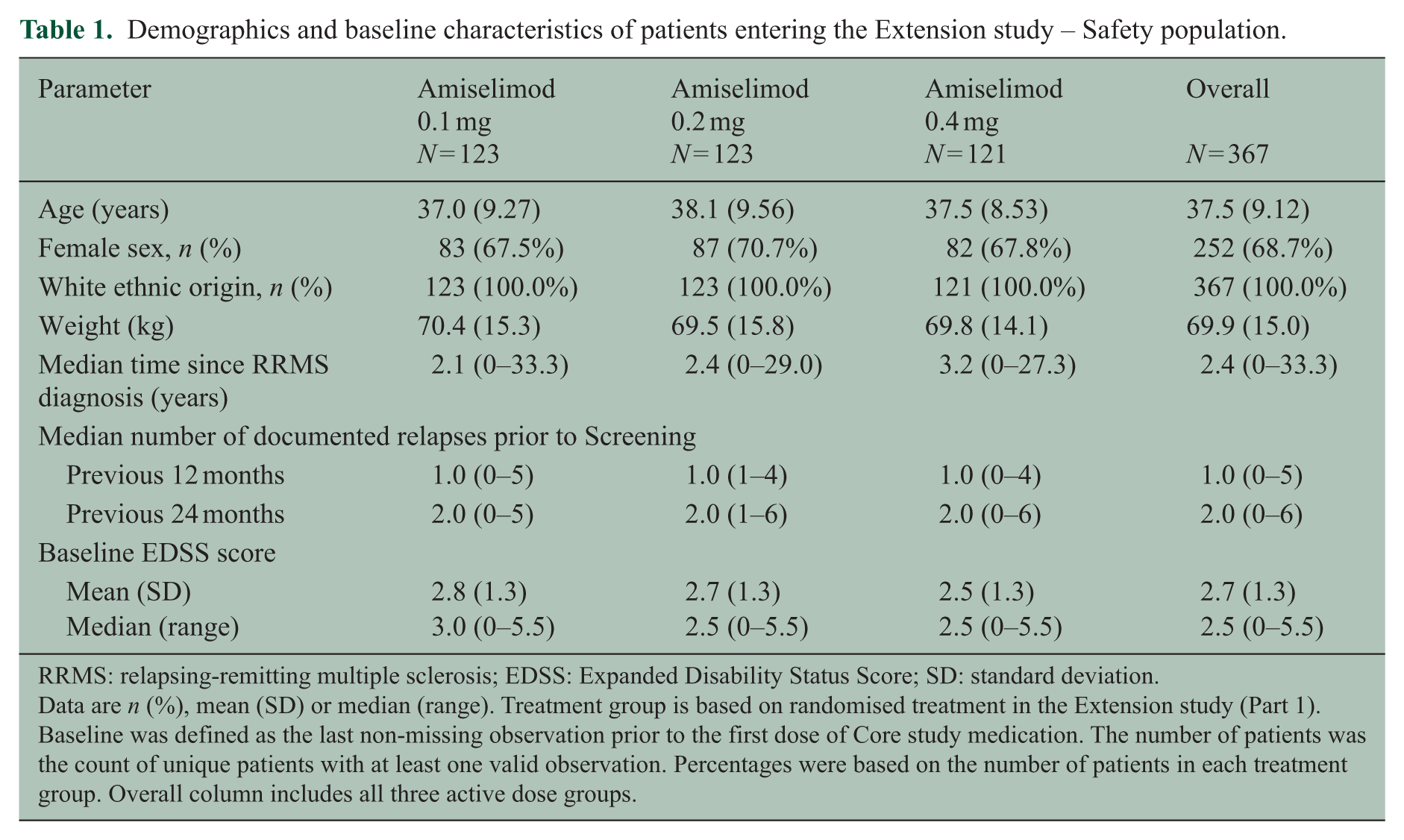
RRMS: relapsing-remitting multiple sclerosis; EDSS: Expanded Disability Status Score; SD: standard deviation.
Data are n (%), mean (SD) or median (range). Treatment group is based on randomised treatment in the Extension study (Part 1). Baseline was defined as the last non-missing observation prior to the first dose of Core study medication. The number of patients was the count of unique patients with at least one valid observation. Percentages were based on the number of patients in each treatment group. Overall column includes all three active dose groups.
Overview of treatment-emergent adverse events – Safety population.
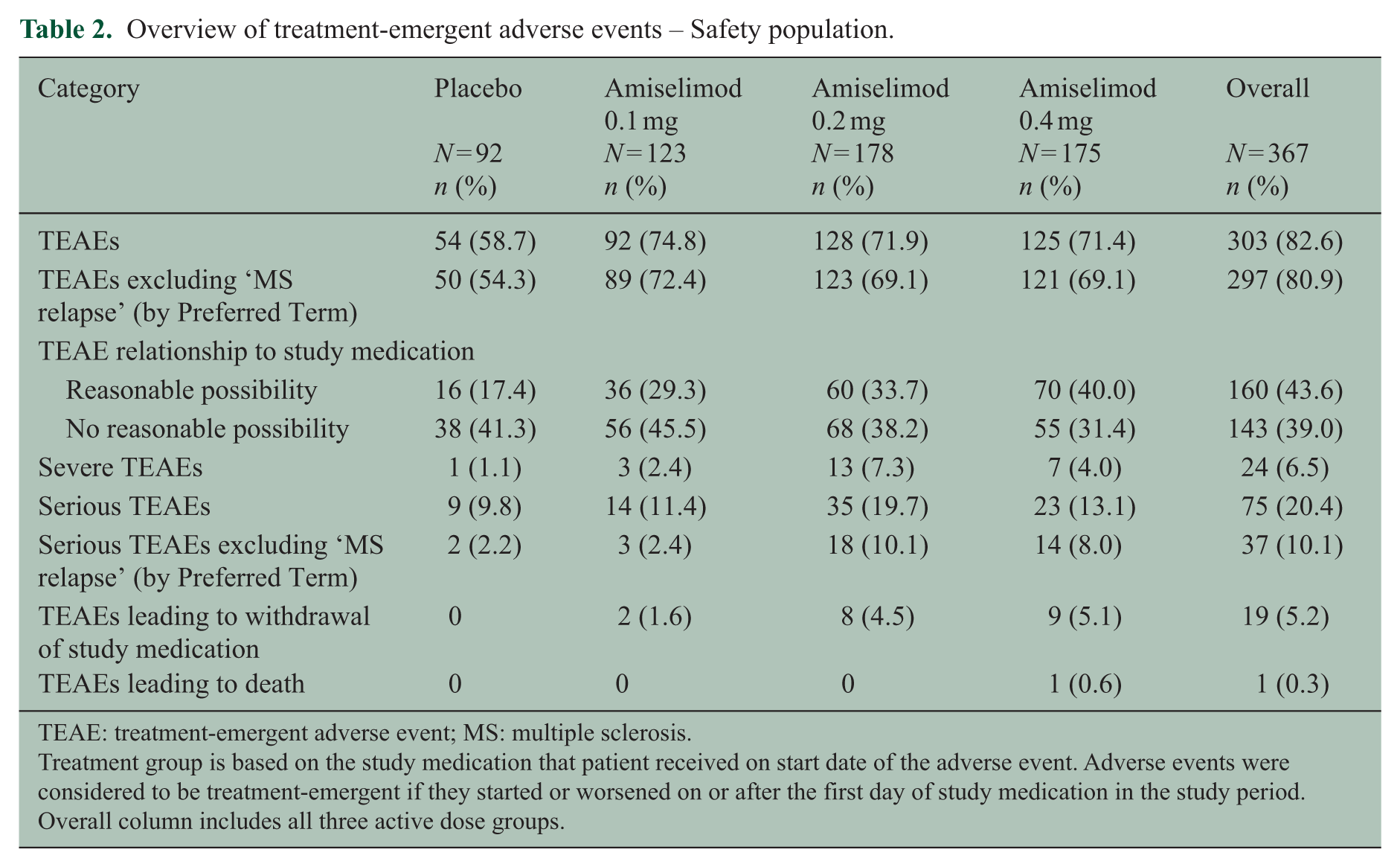
TEAE: treatment-emergent adverse event; MS: multiple sclerosis.
Treatment group is based on the study medication that patient received on start date of the adverse event. Adverse events were considered to be treatment-emergent if they started or worsened on or after the first day of study medication in the study period. Overall column includes all three active dose groups.
Safety and tolerability
Overall (‘Safety population’) analysis
An overview of treatment-emergent adverse events (TEAEs) reported during the study is presented in Table 2. Of the 367 patients who entered the Extension, 303 (82.6%) reported one or more TEAEs. Common TEAEs (>10% overall) were ‘headache’ (62 (16.9%) patients), ‘lymphocyte count decreased’ (62 (16.9%)), ‘nasopharyngitis’ (58 (15.8%)) and ‘MS relapse’ (54 (14.7%)). Overall, 75 (20.4%) patients reported a serious TEAE, of which ‘MS relapse’ was the most common (43 (11.7%)). TEAEs leading to discontinuation of study medication were reported in 19 (5.2%) patients. ‘Lymphocyte count decreased’ (9 (2.5%)) was the most common single reason for withdrawal, but only four (1.1%) patients met the protocol-defined withdrawal criterion of lymphopenia (i.e. absolute lymphocyte count <0.20 × 109/L confirmed in a subsequent re-test) during the Extension study. No case of serious opportunistic infection, macular oedema or malignancy was reported.
A sudden death (recorded as ‘coronary artery insufficiency’ TEAE) was reported during the Extension Part 2 while the patient (24-year-old female MS patient with no other relevant medical or family history) was on treatment with amiselimod 0.4 mg for 5 months (161 days). In the opinion of the investigator, this serious TEAE was not related to study medication. At the post-mortem examination, the coroner concluded that the cause of death was ‘acute coronary failure’ as a result of spasm within the coronary vessels. However, an independent expert cardiac pathologist’s opinion was that it was a negative autopsy without any structural abnormalities of the heart or other organs from which the cause of death was not apparent and interpreted the findings as compatible with the assumption of a Sudden Cardiac Arrhythmic Death Syndrome. 10 The DSMB agreed with this assessment and considered the cause of death as ‘unknown’ (Please see Supplementary Document for more details).
‘By Treatment Sequence’ analysis
A summary of commonly reported TEAEs and serious TEAEs is shown in Table 3. The incidence of TEAEs was not notably different between the two continuous treatment groups (i.e. ‘0.2-mg only’ and ‘0.4-mg only’), except for more frequent incidents of ‘lymphocyte count decreased’ in the 0.4-mg only group (24 (26.1%) versus 16 (17.8%) in the 0.2-mg only group). During the 2-year treatment with amiselimod, three (3.3%) patients in the 0.2-mg only and four (4.3%) in the 0.4-mg only groups were withdrawn due to this event; none of these was associated with clinical signs of infections. The incidence of serious TEAEs (excluding ‘MS relapse’) was lower in the 0.4-mg only group (8 (8.7%)) compared with the 0.2-mg only group (12 (13.3%)). The switch from placebo to active treatment was not associated with any unexpected TEAEs in the placebo-switched groups (i.e. ‘placebo − 0.2 mg’ and ‘placebo − 0.4 mg’).
Summary of TEAEs and serious TEAEs a – ‘By Treatment Sequence’ analysis.
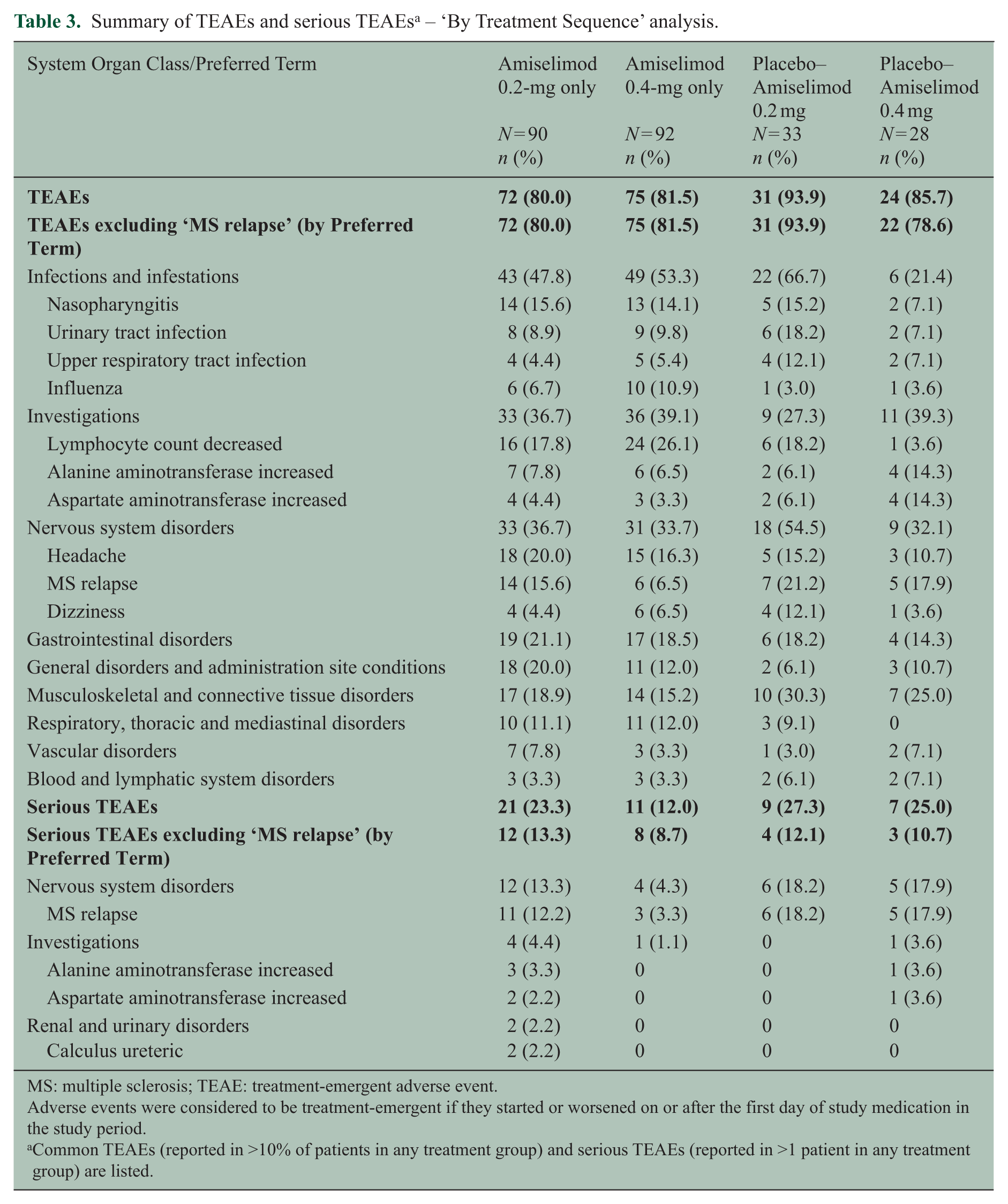
MS: multiple sclerosis; TEAE: treatment-emergent adverse event.
Adverse events were considered to be treatment-emergent if they started or worsened on or after the first day of study medication in the study period.
Common TEAEs (reported in >10% of patients in any treatment group) and serious TEAEs (reported in >1 patient in any treatment group) are listed.
The majority of safety laboratory parameters were within the reference ranges in all four treatment groups and did not show any notable trends over 96 weeks of treatment. Mean values of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were mostly normal and no notable differences were observed between the continuous treatment groups (Supplementary Figure 1). Mean values of gamma-glutamyl transferase (GGT) were slightly increased over time, which was indicative of a dose-dependent trend. There were no reports of clinical symptoms related to any of the abnormal values. Mean neutrophil counts were slightly decreased during the first 12 weeks of dosing, but stabilised thereafter. A total of four patients (1 (1.1%) in the 0.2-mg only and 3 (3.3%) in the 0.4-mg only groups) showed low neutrophil counts (<1.0 × 109/L) at least at one post-dose time-points, but the values returned to normal despite ongoing amiselimod treatment.
Overall, there were no consistent trends in the change from baseline in any of the 12-lead ECG parameters, including HR, and PR, RR, QRS, QT and QTcF intervals (Supplementary Figure 2). Mean systolic and diastolic blood pressure as well as pulse rate were also stable over 96 weeks in all treatment groups.
‘Duration of Exposure’ analysis
Mean hourly HRs derived from Screening (baseline), Day 1, Day 2, Week 4 and Week 24 Holter ECGs were evaluated in three amiselimod dose groups (i.e. 0.1, 0.2 and 0.4 mg; Supplementary Figure 3). Post-dose mean hourly HRs were slightly decreased during the daytime period (8:00–20:00 hours) in all three groups and did not recover to the baseline level within 24 weeks of dosing. However, as mean hourly HRs in this period were consistently higher than 74 bpm in all groups, this magnitude of HR reduction was not considered to be clinically significant. In contrast, there was little HR reduction during the night time period (20:00–8:00 hours). Thus, there was no clinically relevant difference in HR between these groups. The circadian rhythm of HR was well preserved in all three amiselimod doses.
There were no notable differences in the incidence of any pre-defined arrhythmia criterion (including bradycardia, atrioventricular (AV) blocks and ventricular tachycardia) between these groups (Supplementary Table 1). No arrhythmia of clinical concern was observed during the study. Thus, there were no safety concerns raised with respect to potential cardiac effects of amiselimod. The independent review of Holter ECG data by the DSMB cardiologist (E.A.) also confirmed that there were no clinically significant findings throughout the Core and Extension studies.
PD and efficacy
‘By Treatment Sequence’ analysis
During the 96-week treatment period, the mean nadir lymphocyte count achieved was 0.67 × 109/L and 0.56 × 109/L for the 0.2-mg only and 0.4-mg only groups (Figure 2(a)). In the placebo-switched groups, lymphocyte counts were decreased to similar levels to the corresponding doses in the continuous treatment groups after switching to active treatment. Mean lymphocyte counts returned to normal (>1.4 × 109/L) at the Safety Follow-up visit in all groups.
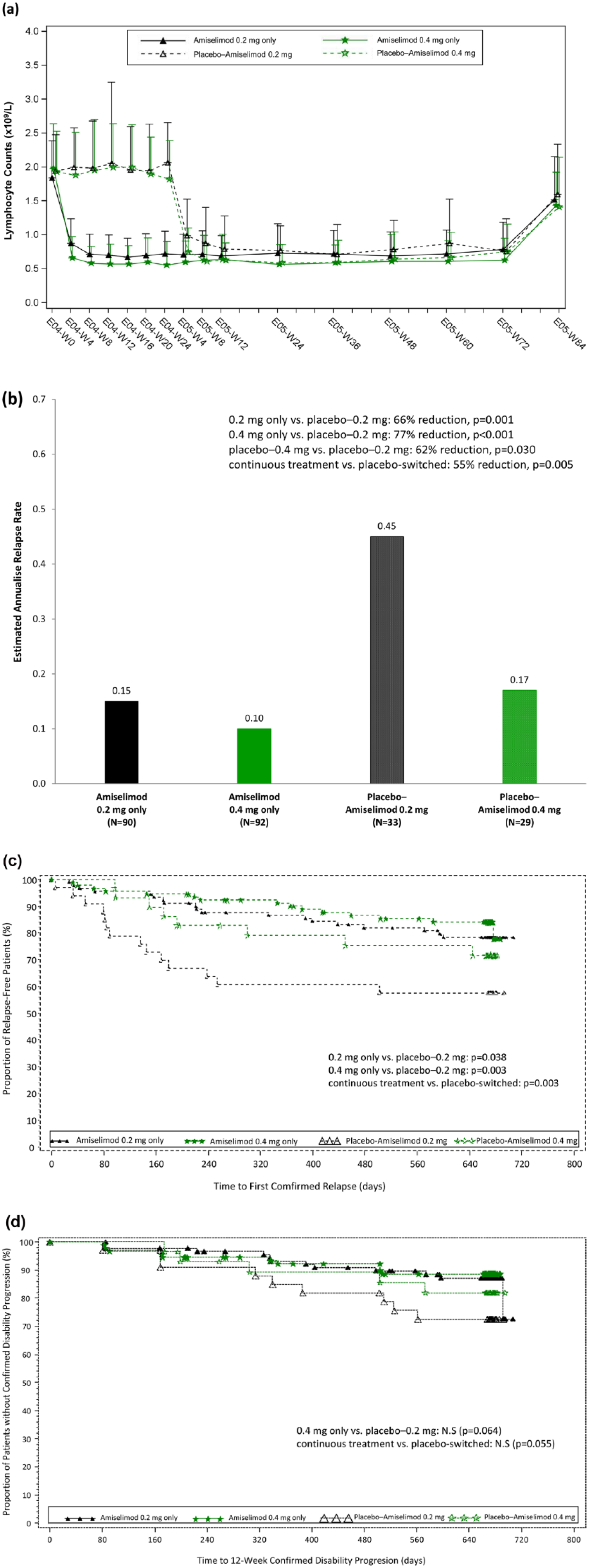
Pharmacodynamics and clinical outcomes up to 96 weeks of treatment – ‘By Treatment Sequence’ analysis. (a)
By the end of 96-week treatment, the ARR was numerically lower in the 0.4-mg only group (0.10) versus the 0.2-mg only group (0.15) (p = 0.259) (Figure 2(b)). In comparison with the placebo-switched groups, the ARR was lower in the continuous treatment groups (p = 0.005) (Figure 2(b)). In addition, time to first confirmed relapse was longer for patients in the continuous treatment groups compared with those in the placebo-switched groups (p = 0.003) and the proportion of relapse-free patients remained higher in the continuous treatment groups at the end of the Extension study (Figure 2(c)).
Time to 12-week confirmed disability progression in the continuous treatment groups was numerically longer compared with the placebo-switched groups (p = 0.055) (Figure 2(d)). There were no notable differences between the treatment groups in the change of EDSS, MSFC or MSQOL-54 scores from baseline (data not shown).
‘Duration of Exposure’ analysis
During 48 weeks of treatment, mean lymphocyte counts were decreased in a dose-dependent manner, achieving reductions from baseline of 40.6%, 63.1% and 70.1% in the amiselimod 0.1-mg (1.11 × 109/L), 0.2-mg (0.67 × 109/L) and 0.4-mg (0.56 × 109/L) groups.
In line with the PD effect, the ARR was decreased dose-dependently in the amiselimod 0.1-mg (0.17), 0.2-mg (0.13) and 0.4-mg (0.05) groups: The ARR in the amiselimod 0.4-mg group was lower compared with the 0.1-mg group (p = 0.020), whereas the difference between 0.2- and 0.4-mg groups was not significant (p = 0.085).
A dose-dependent effect was also observed on MRI-related outcomes (number of Gd-enhanced T1-weighted lesions, and new or enlarged T2-weighted lesions) over 48 weeks of treatment (Supplementary Figure 4a). The proportion of patients free of Gd-enhanced T1-weighted lesions remained higher over time, most visibly in the 0.2-mg (81.4%) and 0.4-mg (83.5%) groups (Supplementary Figure 4a). There were no notable differences in the change in total brain volume across the three groups, whereas the decrease in grey matter volume was lower with increasing dose, indicating a dose-response relationship (Supplementary Figure 4b).
Discussion
Considering the chronic nature of RRMS, the long-term safety and efficacy assessment of disease-modifying therapies are of particular relevance. 1 The MOMENTUM Extension study showed that amiselimod at doses up to 0.4 mg was generally well tolerated when administered in patients with RRMS for up to 2 years. These results were consistent to the findings of the 24-week Core study. 3 There was no clear sign or signal that a long-term use of amiselimod would increase the risks of clinically significant infections (including opportunistic infections), bradyarrhythmia, hepatic and pulmonary dysfunction, macular oedema or malignancy, although the relevant monitoring may still be recommended.
The large proportion of patients who entered the Extension study continued until the end of the 96-week treatment period, supporting a favourable safety and tolerability profile of amiselimod in patients with RRMS. Over 96 weeks of treatment, ‘lymphocyte count decreased ’, a PD effect resulting from the mode of action of amiselimod, was the only TEAE showing a dose-dependent trend between 0.2- and 0.4-mg doses. No serious opportunistic infection or malignancy was reported. A slight dose-dependent increase in mean values of ALT, AST and GGT was observed during the 24-week Core study. 3 Although GGT indicated a dose-dependent trend over time, mean values of ALT and AST were generally stable for both 0.2- and 0.4-mg doses during the Extension. Mean neutrophil counts were slightly decreased, but remained within the reference range throughout the Extension for all groups.
The cardiac safety profiles of the three active doses of amiselimod (0.1, 0.2 and 0.4 mg) were evaluated by Holter ECG over 24 weeks of treatment. A slight decrease in mean hourly HR was observed on all post-dose Holter assessment days in all three dose groups and did not fully recover to the baseline level within 24 weeks. However, a HR reduction of clinical relevance was not seen at any time period in patients treated with amiselimod. The comprehensive arrhythmia analysis did not identify any abnormal finding of clinical concern, which further confirmed the benign cardiac profile of this compound. In view of this overall picture, it appears justified to interpret the fatal TEAE in a patient on amiselimod 0.4 mg as unrelated to the drug although – lacking other identifiable cause – a causal relationship with study medication cannot be fully excluded. The transient T-wave inversion on ECG, exclusively observed at 4–6 hours after the first dose of amiselimod 0.1 mg and then again, several months later at 2–8 hours after the first dose of 0.4 mg would be compatible with a cardiac risk profile specific to this patient unmasked by amiselimod. Of note, the fatal TEAE occurred 5 months after the transient T-wave inversion had been documented. Nevertheless, such phenomena, even if only observed during the initiation of treatment with S1P receptor modulators, should lead to the consideration of further specialist cardiologic work-up.
The ARR was low in both 0.2-mg (0.15) and 0.4-mg (0.10) only groups over 96 weeks of treatment, corresponding well to the significant and dose-dependent decrease in peripheral lymphocyte counts (mean nadir 0.67 × 109/L in the 0.2-mg and mean nadir 0.56 × 109/L in the 0.4-mg). Results were consistent to the findings in the ‘Duration of Exposure’ analysis where a dose-dependent effect on PD, ARR and MRI-related outcomes was observed for up to 48 weeks of treatment. Although a parallel placebo-control group was lacking in the Extension, these dose-dependent effects support a sustained efficacy of amiselimod in RRMS.
Patients who switched from placebo to amiselimod in the Extension study showed a clear improvement in clinical outcomes as compared to the Core study. However, the overall lower ARR, the longer time to first confirmed relapse, the higher proportion of relapse-free patients and the longer time to 12-week confirmed disability progression in the continuous treatment groups versus the placebo-switched groups provide evidence in favour of early and continuous therapy. Similar findings were reported for other immunomodulators; for instance, in the extension studies with fingolimod, an improvement in clinical and MRI-related outcomes was also clearly apparent following the switch from placebo to active treatment, but patients switching after the core studies did not fully catch up with those in the continuous fingolimod treatment groups.11,12
Conclusion
Once-daily oral amiselimod provided a continuous and dose-dependent beneficial effect on clinical and MRI-related outcomes in patients with RRMS. There was no indication that these effects would decrease over time. The improvement observed in patients after switching from placebo to active treatment further supports the value of this treatment, but does also underline the importance of early intervention with effective doses (0.2 and 0.4 mg) of amiselimod for controlling disease activity in RRMS over the longer term.
Given the similar safety profile of the two effective doses and the higher efficacy observed at the 0.4 mg dose, amiselimod 0.4 mg appears to be the dose to further study in this indication.
Footnotes
Acknowledgements
The authors thank the patients and their families, the MOMENTUM study investigators and coordinators and all the staff at the study sites. The authors also thank Professor Mary N Sheppard (Cardiovascular Pathology Unit, St George’s, University of London, London, UK) and PD Dr Michael Kühne (Cardiology, Department of Medicine, University Hospital Basel, Basel, Switzerland) for scientific expert advice, Professor Bernd C Kieseier (Heinrich-Heine University, Düsseldorf, Germany) for his helpful suggestions in study design and Ms Kristin Greenough (served as clinical operational lead of Mitsubishi Tanabe Pharma Europe while the study was conducted) for overseeing the study. All authors had a full access to the study data and reviewed and approved the manuscript. The corresponding author has final responsibility for the decision to submit for publication. L.K., as chair of this study’s Steering Committee, was involved in drafting and reviewing the study protocol, overseeing the conduct of the study and reviewing statistical analysis. L.K. was also involved in and contributed to the writing and review of the manuscript and, as a corresponding author, had final responsibility for the decision to submit for publication. D.L.A. participated in study design, MRI data analysis and review and approval of the manuscript. A.B.O., T.D. and T.S. participated in study design, data analysis and review and approval of the manuscript. A.J.C. participated in study design, cardiovascular data analysis and review and approval of the manuscript. M.D. (served as scientific director), A.P. (project manager), P.N. (lead statistician) and T.H. (global medical lead) all participated in study design, drafting and reviewing the study protocol and data analysis. M.D., A.P. and P.N. reviewed and approved the manuscript. T.H. drafted, reviewed and approved the manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: L.K.’s institution (University Hospital Basel) received funding in the last 3 years and used exclusively for research support: steering committee, advisory board and consultancy fees (Actelion, Addex, Bayer Health Care, Biogen, Biotica, Genzyme, Lilly, Merck, Mitsubishi Tanabe Pharma, Novartis, Ono Pharma, Pfizer, Receptos, Sanofi-Aventis, Santhera, Siemens, Teva, UCB, Xenoport); speaker fees (Bayer Health Care, Biogen, Merck, Novartis, Sanofi-Aventis, Teva); support of educational activities (Bayer Health Care, Biogen, CSL Behring, Genzyme, Merck, Novartis, Sanofi, Teva); royalties for Neurostatus products; grants (Bayer Health Care, Biogen, Merck, Novartis, Roche, Swiss MS Society, the Swiss National Research Foundation, the European Union, Roche Research Foundations). D.L.A. reports personal fees for consulting from Acorda, Biogen, Hoffman LaRoche, MedImmune, Mitsubishi Tanabe Pharma, Novartis, Receptos, Sanofi-Aventis; grants from Biogen and Novartis; and an equity interest in NeuroRx Research which was the image analysis centre for the trial. A.B.O. reports personal fees for consulting from Biogen, Hoffman LaRoche, Genentech, MedImmune, Mitsubishi Tanabe Pharma, Novartis, Receptos, Sanofi-Genzyme; and grants from Novartis and Sanofi-Genzyme. A.J.C. reports personal fees as an adviser/speaker for: Actelion, AstraZeneca, Bayer, Biotronik, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, ChanRX, Daiichi, Gilead, GlaxoSmithKline, InfoBionic, Incarda, Johnson and Johnson, Medtronic, Menarini, Merck, Mitsubishi Tanabe Pharma, Novartis, Otsuka, Pfizer, Sanofi, Servier, St. Jude Medical, Takeda and Xention. T.D. serves on scientific advisory boards/steering committees for Novartis, Merck Serono, Biogen Idec, GeNeuro, Genzyme, Mitsubishi Tanabe Pharma, TEVA Pharma, Octapharma and Bayer Schering Pharma; has received funding for travel and/or speaker honoraria from Biogen Idec, Novartis, Genzyme, Merck Serono and Bayer Schering Pharma; and receives research support from Novartis, Biogen Idec, the Swiss National Research Foundation, the European Union and the Swiss MS Society. T.S.’s current (DKD Helios Klinik Wiesbaden) or previous institution (University Hospital Basel) has, in the last 3 years, received fees for consulting and speaking from Mitsubishi Tanabe Pharma, Biogen Idec, Desitin, Novartis, Actelion, Sanofi-Genzyme, TEVA and Electrocore. T.S. received research grants from the Swiss MS Society, Novartis, EFIC-Grünenthal grant and Swiss National Science Foundation. M.D., A.P., P.N. and T.H. are employees of Mitsubishi Tanabe Pharma Europe (European subsidiary of Mitsubishi Tanabe Pharma Corporation).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by the Mitsubishi Tanabe Pharma Corporation. The sponsor participated in the design of the study, conduct of the study, data management, data analysis and interpretation and preparation, review and approval of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.