
Abstract
Recent advancements in cancer multi-omics have transformed our understanding of cancer biology by integrating genomics, transcriptomics, proteomics, and metabolomics. These integrative approaches have led to the identification of novel biomarkers and therapeutic targets, offering deeper insights into the molecular intricacies of various cancers, including breast, lung, gastric, pancreatic, and glioblastoma. Despite these advances, challenges remain, such as the integration of disparate data types and the interpretation of complex biological interactions. However, developments in proteogenomics and mass spectrometry have enhanced the correlation between molecular profiles and clinical features, refining the prediction of therapeutic responses. Future research in cancer drug discovery is poised to benefit from multi-omics approaches, improving the precision and efficacy of personalized therapies. By developing integrative network-based models, researchers aim to address challenges related to heterogeneity, reproducibility, and data interpretation. A standardized framework for multi-omics data integration could revolutionize cancer research, optimizing the identification of novel drug targets and enhancing our understanding of cancer biology. This complete approach holds the promise of advancing personalized therapies by fully characterizing the molecular landscape of cancer, ultimately improving patient outcomes through more effective and targeted treatment strategies. This narrative review underscores the potential of multi-omics approaches to transform cancer research and improve patient outcomes through more precise and effective treatments.
Keywords
Introduction
The advent of large-scale molecular profiling methods, collectively known as omics technologies, has revolutionized our understanding of disease mechanisms. These methods have uncovered novel pathways, disease-associated loci, biomarkers, and therapeutic targets. 1 While early focused on single-omics layers, such as the genome or transcriptome, it has become increasingly clear that a comprehensive understanding of disease and normal physiology requires integrative, multi-omics analyses. Biological systems operate through complex, interconnected layers, including the genome, transcriptome, proteome, metabolome, microbiome, and lipidome. Genetic information flows through these layers to shape observable traits, 2 and elucidating the genetic basis of complex phenotypes demands an analytical framework that captures these dynamic, multi-layered interactions. 3 Integrating diverse omics datasets presents substantial computational challenges. Addressing these requires advanced statistical, network-based, and machine learning methods to model interdependencies and extract meaningful biological insights. 4 Among these, network-based approaches offer a powerful lens for analyzing multi-omics data (Figure 1). By modeling molecular features as nodes and their functional relationships as edges, these frameworks capture complex biological interactions and can identify key subnetworks associated with disease phenotypes. 5 Furthermore, many network-based techniques an incorporate prior biological knowledge, enhancing interpretability and predictive power. As a result, they have gained prominence in multi-omics research, particularly in elucidating disease mechanisms and informing drug discovery. 6 Despite growing interest, most prior reviews have centered on either single-omics network analyses or broad overviews of multi-omics integration. 7 In this narrative review, we aim to bridge that gap by highlighting the role of network-based strategies in multi-omics integration, with a focus on their transformative potential in cancer research and precision medicine.

Network of connected nodes to operate the molecular features for cancer research.
Navigating Cancer Complexity: Integrative Multi-Omics Methodologies for Clinical Insights
The advancement of multi-omic technologies has transformed the landscape of cancer research, providing unprecedented insights into the molecular basis of cancer. This comprehensive approach integrates data from various omics fields, including genomics, transcriptomics, proteomics, metabolomics, and lipidomics, thus offering a holistic view of the molecular landscape of cancer (Table 1). This integrated approach facilitates the identification of novel therapeutic targets and the development of personalized treatment strategies tailored to the unique molecular profile of each patient’s tumor.
Comparison of multi-omics approaches: differences, advantages, and limitations.

Abbreviations: CNV, copy number variation; SNP, single-nucleotide polymorphism.
Genetic and genomic variations
Mutations
Mutations in cancer are broadly categorized into driver mutations and passenger mutations. Driver mutations are changes in the genome that provide a growth advantage to cells and are directly involved in the oncogenic process. These mutations are critical for cancer development and progression. They typically occur in genes involved in key cellular processes such as cell growth regulation, apoptosis (programmed cell death), and DNA repair. Updated technologies such as next-generation sequencing (NGS), by enabling the comprehensive analysis of an entire genome, exome, or transcriptome, have revolutionized cancer research by allowing scientists to identify numerous cancer-associated mutations with high accuracy. 1 This technology has provided profound insights into the molecular mechanisms of cancer and has significantly advanced our understanding of tumor biology and potential targets for therapy. For example, mutations in the TP53 gene, which encodes a tumor suppressor protein, are found in approximately 50% of all human cancers, highlighting its crucial role in maintaining cellular integrity. 2
Copy number variations
Copy number variations (CNVs) represent an unexpectedly frequent, dynamic, and complex form of genetic diversity. CNVs involve duplications or deletions of large regions of DNA, leading to variations in the number of copies of genes. These variations can significantly influence cancer development by altering gene dosage, which represents the number of copies of a particular gene present in a cell. They can lead to the overexpression of oncogenes, whose mutation or aberrantly high expression contributes to cancer development. Conversely, CNVs can also result in the under expression of tumor suppressor genes, which normally help prevent cells from becoming cancerous. Both scenarios can contribute to the initiation and progression of cancer. 3
A well-known example of a CNV influencing cancer is the amplification of the human epidermal growth factor receptor 2 (HER2) gene. The HER2 gene plays a role in the growth and division of cells. In about 20% of breast cancers, the HER2 gene is amplified, leading to the overexpression of the HER2 protein on the surface of cancer cells. This overexpression is associated with aggressive tumor behavior and a poor prognosis, as it promotes rapid growth and spread of the cancer cells. The identification of HER2 gene amplification has had significant clinical implications. It led to the development of targeted therapies such as trastuzumab, a monoclonal antibody that specifically targets the HER2 protein thus inhibiting the growth of cancer cells. This targeted approach has significantly improved outcomes for patients with HER2-positive breast cancer. The development of HER2-targeted therapies underscores the importance of understanding CNVs and their impact on gene expression in cancer. It highlights how genetic research can lead to more precise and effective treatments, offering hope for better management and potential cures for various cancers. 4
Single-nucleotide polymorphisms
Single-nucleotide polymorphisms (SNPs) are the most common type of genetic variation among people. These variations can occur in coding regions of genes, non-coding regions, or in the intergenic regions between genes. While most SNPs have no effect on health or development, some can affect how cancers develop or respond to drugs. Single-nucleotide polymorphisms can increase an individual’s susceptibility to cancer by affecting genes involved in critical cellular processes such as DNA repair, cell growth, and apoptosis. 5 Thus, SNPs influence cancer risk by altering gene expression or the activity of proteins. For example, SNPs in the BRCA1 and BRCA2 genes significantly increase the risk of developing breast and ovarian cancers. Pharmacogenomics studies have also used SNP data to predict patient responses to cancer therapies, improving treatment efficacy and reducing toxicity. 6 Single-nucleotide polymorphisms in genes encoding enzymes involved in drug metabolism can significantly influence the effectiveness and toxicity of chemotherapeutic agents. These genetic variations can lead to differences in the response to chemotherapy drugs. For example, certain SNPs might result in faster or slower metabolism of a drug, altering its concentration in the bloodstream and consequently its therapeutic effect. In addition, these variations can affect the likelihood of adverse reactions, as the altered metabolism might produce toxic metabolites or affect the drug’s ability to target cancer cells effectively. Understanding these genetic differences is crucial for personalized medicine, allowing health care providers to tailor chemotherapy treatments to achieve the better outcome for patients while minimizing harmful side effects. 7
In addition, certain SNPs are associated with cancer prognosis by predicting disease outcome and tailor treatment strategies accordingly. A specific SNP in TP53 (eg, rs1042522) can predict a poorer prognosis in several cancers, including breast and lung cancers. Patients with certain alleles of this SNP may have a higher risk of tumor progression and a lower survival rate.8-10 A better knowledge of SNPs associated with patient prognosis may lead to more intensive monitoring and the use of combination therapies.
Integrating genetic and genomic data
The integration of data from genetic and genomic variations with other omic data is critical for a comprehensive understanding of cancer. Genetic and genomic variations, such as single SNPs, insertions, deletions, and CNVs, provide crucial insights into the hereditary and somatic alterations that drive cancer development and progression. However, to fully understand the complexity of cancer biology, it is essential to combine this genetic and genomic information with other layers of omic data, such as transcriptomics (gene expression profiles) and proteomics (protein expression and modifications).
For instance, combining mutational data with transcriptomic data helps to identify which genes are not only mutated but also aberrantly expressed in cancer cells. This integration highlights the activation or suppression of specific signaling pathways that are critical for cancer cell survival and proliferation. Similarly, proteomic data reveal post-translational modifications of proteins, such as phosphorylation, which play a crucial role in regulating protein function and signaling networks within cells.
By examining these data sets together, the pathways that are disrupted in cancer can be elucidated, and key nodes, such as critical proteins or genes within these pathways, that may be targeted with therapeutic drugs can be identified. For example, if a particular signaling pathway is found to be overactive due to both a genetic mutation and increased expression of a pathway component, targeting this pathway with a specific inhibitor could be an effective treatment strategy.
This integrative approach has also significant implications for understanding mechanisms of drug resistance. Cancer cells often develop resistance to therapies through genetic mutations or adaptive changes in gene and protein expression. By integrating genetic, transcriptomic, and proteomic data, the mechanisms of cancer resistance as well as the mechanisms by which cancer cells are resistant to the effects of treatment might be elucidated. This knowledge guides the development of combination therapies that target multiple pathways simultaneously or sequentially, thereby overcoming or circumventing resistance and improving treatment efficacy.11,12
Transcriptomics and epigenomics
Transcriptomics examines RNA transcripts to identify active genes and their expression levels in cancer cells, providing a functional readout of the genetic information. This omic layer is crucial for identifying which genes are upregulated in cancer and may be potential targets for therapy. Techniques like RNA-seq not only quantify gene expression but also detect novel transcripts, splice variants, and non-coding RNAs that could play roles in cancer.11,13
Epigenomics, particularly the study of DNA methylation, offers insights into gene regulation mechanisms that do not alter the DNA sequence but still affect gene expression. Altered DNA methylation patterns are frequently observed in cancer and are commonly linked to the silencing of genes, especially tumor suppressor genes. By integrating methylation data with genomic and transcriptomic information, researchers can gain deeper insights into the molecular underpinnings of cancer and potentially identify novel avenues for the development of more effective epigenetic therapies. 14
Proteomics, lipidomics, and metabolomics
Proteomics provides insights into the proteome, identifying protein expressions, modifications, and interactions that are crucial for cancer progression. This data is invaluable for discovering new drug targets and understanding drug resistance mechanisms. Advanced mass spectrometry techniques have enabled the quantitative and qualitative analysis of the proteome in complex biological samples, revealing protein-level alterations associated with cancer. 15
Lipidomics examines the role of lipids in cancer, which are critical not only as structural components of cell membranes but also in signaling pathways that regulate cell proliferation, migration, and death. Changes in lipid metabolism have been linked to cancer aggressiveness and therapy resistance. 16
Metabolomics, the study of metabolic alterations, is crucial for understanding how changes in metabolic pathways support cancer growth and survival. Metabolomic profiling can reveal energy production changes and dependencies on specific metabolites, offering potential targets for therapy that could starve cancer cells by cutting off their energy supply.3,17
Integrative multi-omic analysis
Integrating data from these diverse omic streams is a significant challenge that requires advanced computational tools and sophisticated bioinformatics approaches. This integration allows for the construction of a comprehensive model of cancer biology, accounting for the interactions between the genome, epigenome, transcriptome, proteome, lipidome, and metabolome. Network and systems biology approaches are particularly useful in this context. They enable the modeling of complex biological interactions and the identification of key nodes and pathways that could be targeted therapeutically. Such models not only predict how different components of the cancer cell interact but also how they might respond to various therapies. The understanding how genes and proteins interact within these networks, enables the identification of potential targets for new drugs or repurpose existing drugs to disrupt cancer progression effectively. In addition, integrating these data streams supports the comprehension of the dynamic changes that occur in cancer cells over time and in response to pharmacological treatment, thus providing insights into mechanisms of drug resistance and disease relapse.
Furthermore, these integrative models support personalized medicine approaches by predicting the most effective therapy for each patient based on the unique molecular cancer features. This comprehensive view of cancer biology is critical for developing more effective treatment strategies and improving patient outcomes. 11
Computational Frameworks for Multi-Omics Studies
Recent progress in high-throughput sequencing technologies has enabled the simultaneous measurement of multiple molecular features in cancer within a single experimental setting. These techniques allow for rapid and unbiased profiling of somatic mutations, copy number variations (CNVs), and the expression of mRNA, non-coding RNAs, and proteins. To interpret such complex data, a variety of computational algorithms have been developed for multi-omics clustering, aiming to extract coherent patterns from heterogeneous data sources. 18 These methods have proven essential in delineating clinically relevant subtypes of complex diseases such as cancer. The integration of multi-omics data from the same patient samples allows researchers to uncover intricate biological structures that may not be evident when analyzing a single data modality. 19 For instance, cancer subtypes can be more precisely defined by combining gene expression profiles with mutation data from the same individuals. Multi-omics clustering mitigates the limitations and potential biases inherent in single-omics analyses, as the inclusion of multiple molecular layers yields a more comprehensive and robust representation of cellular states, spanning from genomic to epigenomic levels. 20
Various tools have been developed to analyze multi-omics datasets with specific objectives and goals: (1) to identify disease subtypes or classify subgroups, (2) to pinpoint potential biomarkers for diagnostics and driver genes for diseases, and (3) to gain insights into disease biology. 21 Most multi-omics frameworks are based on Bayesian statistics, similarity networks, joint non-negative matrix factorization, and sparse canonical correlation analysis.14,22-24 Several multi-omics tools are widely used in the medical field and demonstrate superior performance in subtype prediction and survival analysis. However, these tools are based on different mathematical theories and support varying ranges of data types. The performance of multi-omics integration methods can differ markedly, even when applied to identical datasets, due to variability in the underlying biological characteristics of the study population. 5 Consequently, extracting meaningful biological insights from multi-omics data remains a complex challenge, both computationally and biologically. This underscores the importance of carefully selecting appropriate analytical tools tailored to the specific context of each study.
Newest and most promising multi-omics approaches
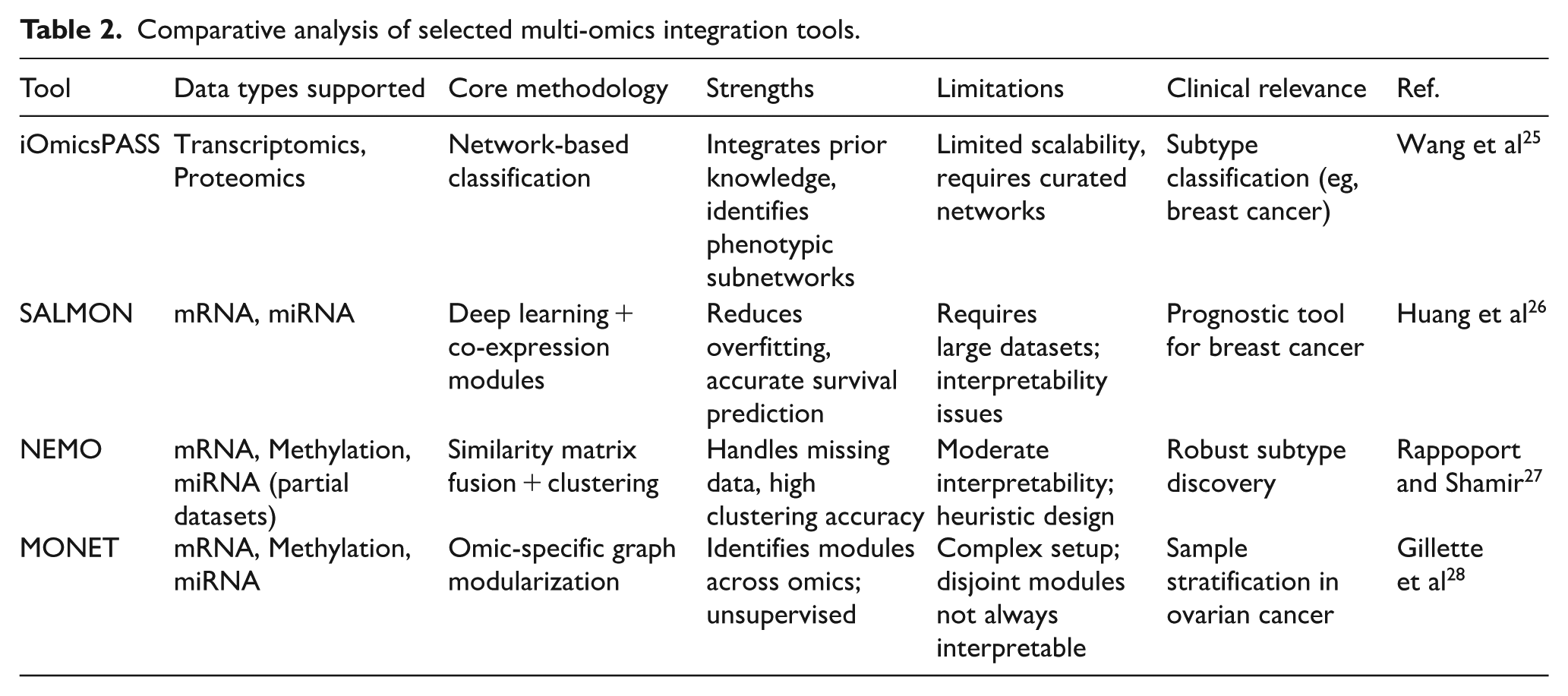
Recent advancements in multi-omics approaches have introduced several innovative methods that hold great promise for the integration and analysis of complex biological data. Four such cutting-edge techniques—iOmicsPASS, SALMON, NEMO, and MONET—are particularly noteworthy due to their advanced computational frameworks and their ability to provide deeper insights into cancer biology.
iOmicsPASS
iOmicsPASS is a network-based analytical framework distinguished by its capacity to integrate genome-informed networks with multi-omics datasets. Introduced by Koh et al in 2019, the algorithm transforms omics data into biological interaction scores, which are subsequently used to construct phenotype-specific networks. These networks are generated using a modified version of the nearest shrunken centroid method. 25 iOmicsPASS has proven particularly effective in refining the classification of breast invasive ductal carcinoma (IDC) subtypes through the joint analysis of mRNA expression and protein abundance. This integrative approach enabled the identification of novel transcriptional regulatory networks unique to specific breast cancer subtypes—networks that remained undetectable using single-omics analyses alone—highlighting the method’s potential to uncover previously unrecognized biological mechanisms.
SALMON
Survival Analysis Learning with Multi-Omics Neural Networks (SALMON) is a deep learning-based method that leverages co-expression networks to process multi-omics datasets. Introduced by Huang et al 26 in 2019, SALMON addresses the issue of overfitting that often arises in multi-omics studies with numerous features but limited. By computing eigengenes from co-expression modules, SALMON effectively reduces overfitting and enhances prediction accuracy. For example, in an analysis of mRNA and miRNA datasets from 583 female patients with breast invasive carcinoma, the SALMON algorithm produced robust predictions of survival outcomes, underscoring its potential utility in clinical settings for cancer prognosis and therapeutic decision-making.
NEMO
Neighborhood-based Multi-Omics Clustering (NEMO), developed by Rappoport and Shamir in 2019, is a multi-omics clustering method known for its ability to handle partial datasets without requiring data imputation. NEMO, developed by Rappoport and Shamir in 2019, is a multi-omics clustering method known for its ability to handle partial datasets without requiring data imputation. NEMO first computes an inter-patient similarity matrix for each omics dataset and then integrates these matrices into a single combined matrix. NEMO first computes an inter-patient similarity matrix for each omics dataset and then integrates these matrices into a single combined matrix. 27 Clusters are identified by calculating patient similarities using an adjusted Rand index based on distance. NEMO has outperformed other multi-omics clustering algorithms, particularly in multi-omics datasets from 10 different cancers. Its capability to improve cluster detection even with partial datasets makes it a robust tool for uncovering meaningful biological patterns in incomplete data.
MONET
Multi-Omic Clustering by Non-Exhaustive Types (MONET) is a computational method developed to identify consistent modules across multiple omics datasets. Introduced by Rappoport et al in 2020, the algorithm integrates 3 types of omics data—mRNA expression, DNA methylation, and miRNA expression—by constructing an edge-weighted graph for each dataset, where nodes represent patient samples and edges quantify similarity between them. 28 MONET subsequently detects disjoint module sets across these omics graphs, grouping patients based on shared molecular features. In a benchmarking study involving 287 patients with ovarian serous cystadenocarcinoma, MONET identified 4 distinct sample modules that correlated with venous invasion status and overall survival. These findings underscore MONET’s utility in uncovering clinically meaningful patient subgroups, thereby supporting its potential role in advancing personalized medicine and targeted therapeutic strategies.
These 4 approaches—iOmicsPASS, SALMON, NEMO, and MONET—represent the forefront of multi-omics data integration and analysis. They address key challenges such as data heterogeneity, overfitting, and incomplete datasets, thereby providing more accurate and comprehensive insights into cancer biology. By leveraging advanced computational techniques like network analysis, deep learning, and clustering, these methods offer significant improvements in identifying disease subtypes, potential biomarkers, and prognostic indicators. 18
As multi-omics research continues to evolve, these approaches are expected to play a critical role in advancing our understanding of complex diseases like cancer. Their ability to integrate and analyze diverse molecular data will be instrumental in developing more effective diagnostic tools, personalized treatments, and targeted therapies, ultimately improving patient outcomes and advancing the field of precision medicine. Table 2 reports a comparative analysis of selected multi-omics integration tools.
Comparative analysis of selected multi-omics integration tools.
A snapshot of challenges integrating multi-omics approaches
Although numerous cancer omics layers are accessible through established databases, the effective integration and interpretation of these data remain major challenges. A key limitation lies in the acquisition of multi-layered datasets, which are often generated using diverse experimental modalities that exhibit distinct statistical characteristics. These differences arise from factors such as genomic heterogeneity among patients, varying cell type compositions, and technical variability. Moreover, each omics layer introduces complex correlation structures and hidden confounding effects (systematic biases), further complicating data integration. 29 For instance, combining proteomics with transcriptomic data presents specific challenges: proteomic analyses typically capture only a subset of the expressed genome and are influenced by technical constraints in sample preparation, as well as biological factors such as post-translational modifications, protein localization, and degradation. These elements hinder accurate modeling of relationships across omics layers. 30
Another critical barrier is the absence of standardized protocols for storing and sharing multi-omics data across cancer repositories. This inconsistency often results in the underutilization of rich molecular datasets. Multi-modal cancer data are frequently hosted on web platforms using differing processing pipelines, normalization methods, and genome reference versions, leading to substantial variability across studies. 31 The lack of unified data formats poses a significant obstacle for researchers seeking to integrate, compare, or reproduce findings from distinct studies. Thus, there is a growing demand for standardized, reproducible data formats in multi-omics cancer research. In addition, the prevalence of missing values across datasets further hinders effective data integration and analysis, representing yet another major bottleneck in the field. 32
While many integration strategies show promise in research settings, translating them to clinical applications poses substantial hurdles:
Batch effects and technical noise: data generated across laboratories or platforms often suffer from variability due to differences in protocols, reagents, and instruments. These batch effects can obscure true biological signals unless rigorously corrected. 19
False positives and overfitting: the high dimensionality of multi-omics data, especially when sample sizes are limited, can lead to overfitting and spurious associations if not properly regularized or validated on independent cohorts. 20
Sample size limitations: especially in rare cancer subtypes, obtaining sufficiently large and well-annotated multi-omics datasets remains a critical bottleneck. This restricts statistical power and generalizability. 33
Missing data across omics layers: Real-world clinical datasets often contain missing values due to limited material, failed assays, or cost constraints. Imputation strategies can help but may introduce bias or uncertainty. 34
Ethical and legal considerations: Integrating patient-derived multi-omics data at scale requires compliance with data privacy regulations (eg, GDPR, HIPAA) and robust informed consent frameworks. These challenges are compounded in multi-institutional or international studies. 35
Addressing these challenges will be crucial for the successful clinical translation of multi-omics tools. Solutions may include the adoption of harmonized pipelines, federated learning models, robust data sharing agreements, and a stronger focus on reproducibility and metadata standards.
Emerging frontiers in multi-omics: spatial omics, single-cell, and data privacy
As integrative multi-omics continues to evolve, emerging technologies are expanding the landscape of biological discovery. Three particularly transformative frontiers, spatial omics, single-cell multi-omics, and privacy-preserving computational strategies are redefining the granularity and scope of multi-omics research.
Spatial Omics technologies enable the measurement of gene expression, protein distribution, and even chromatin accessibility directly within the spatial context of intact tissue sections. Unlike bulk or dissociated-cell sequencing methods, spatial transcriptomics preserves tissue architecture, allowing researchers to link molecular patterns with histopathological features. 36 This is particularly valuable in oncology, where the tumor microenvironment—including immune cell infiltration, angiogenesis, and stromal remodeling—plays a critical role in disease progression and therapeutic resistance. Recent advances, such as Slide-seq, 10x Genomics Visium, and MERFISH, have enhanced spatial resolution and multiplexing capacity, facilitating high-definition tumor mapping at near-single-cell resolution. 37
Single-Cell Multi-Omics approaches integrate multiple data modalities (eg, genome, epigenome, transcriptome, proteome) at the level of individual cells. These techniques are crucial for dissecting cellular heterogeneity, identifying rare subpopulations such as cancer stem cells, and mapping lineage trajectories in tumor evolution. 25 Technologies like scRNA-seq (single-cell RNA sequencing) combined with ATAC-seq or CITE-seq enable simultaneous quantification of gene expression and chromatin accessibility or surface protein levels, respectively. 38 The resulting data reveal cell states and intercellular dynamics that are obscured in bulk analyses. Single-cell methods are also key for understanding mechanisms of immune evasion, therapy resistance, and tumor relapse.
Data Privacy and Federated Learning are emerging as critical considerations in the clinical translation of multi-omics research. 39 Multi-center studies often face regulatory hurdles related to patient data sharing, particularly in regions governed by strict frameworks such as the General Data Protection Regulation (GDPR) or the Health Insurance Portability and Accountability Act (HIPAA). Federated learning (FL) offers a promising solution by enabling machine learning models to be trained across decentralized datasets without transferring sensitive data to a central server. Each participating site retains local control over data, sharing only encrypted model updates. 39 This strategy not only enhances privacy but also facilitates collaboration across institutions and countries, fostering reproducibility and model robustness. In parallel, differential privacy and homomorphic encryption methods are being explored to further safeguard patient-level information while preserving analytical power.
Together, these emerging approaches overcome key limitations of current multi-omics studies, spatial resolution, cellular granularity, and secure data use, enabling more actionable and ethically sound clinical applications.
Multi-Omics Insights Across Major Cancer Types
The application of multi-omics approaches has significantly enhanced our understanding of cancer heterogeneity, resistance mechanisms, and therapeutic vulnerabilities across a range of malignancies. This section synthesizes recent findings from integrative studies on 6 representative cancer types, breast, lung, gastric, pancreatic, glioblastoma, and acute myeloid leukemia (AML). For each type, we summarize the omics layers used, major discoveries, and clinical implications. While differences in methodological depth and dataset availability exist, key cross-cutting insights emerge, including the importance of proteogenomics in therapy response, the role of the tumor microenvironment in immune evasion, and the predictive value of metabolic and epigenomic alterations.
Recent advancements in cancer multi-omics have significantly enhanced our understanding of the molecular underpinnings of various cancers, such as breast, lung, gastric, and glioblastoma, by integrating genomics, transcriptomics, proteomics, and metabolomics. These approaches have identified novel biomarkers and therapeutic targets, providing crucial insights into the molecular and clinical characteristics of cancer patients. For instance, in breast cancer, these analyses have uncovered distinct molecular subtypes, facilitating precise prognostic predictions and tailored therapies. In lung cancer, multi-omics has elucidated the role of specific gene mutations and metabolic pathways in tumor progression, enabling more personalized treatment approaches. Gastric cancer studies have revealed complex interactions between genetic alterations and the tumor microenvironment, offering new avenues for targeted interventions. Glioblastoma research has benefited by mapping tumor cell heterogeneity and identifying key drivers of resistance to conventional therapies. Despite these advances, challenges remain, such as addressing incompatibilities in related biological processes in genomics and transcriptomics. However, recent developments in proteomics, particularly mass spectrometry, have facilitated proteogenomic approaches that integrate genomic data with proteomics and information on post-translational modifications, leading to large-scale studies by Clinical Proteomic Tumor Analysis Consortium (CPTAC).28-31 Multi-omics clustering has refined the correlation between molecular profiles and clinical features, highlighting coherent subtypes across datasets that significantly affect predicting clinical relevance and therapeutic response. These findings collectively enhance our understanding of cancer biology, paving the way for more effective and personalized treatment strategies.
Breast cancer
Multi-omics analyses have significantly advanced our understanding of breast cancer biology and treatment strategies. 32 highlighted the power of multi-omics to address challenges such as multidrug resistance (MDR) and relapse in breast cancer therapy, emphasizing the necessity of understanding multi-omic mechanisms to develop effective treatments. Integrating multi-omic data, which includes prognostic, pathological, and clinical factors, provides valuable insights for therapeutic strategies.
Ektefaie et al introduced weakly supervised deep learning models for the analysis of multi-omics data derived from breast cancer biopsy specimens. 40 These automated frameworks, developed for tumor detection and histopathological subtype classification, exhibited high predictive accuracy and were successfully validated using independent patient cohorts.
In the context of epigenomic and genomic analyses, researchers identified frequent activation of the CT83 gene in triple-negative breast carcinomas (TNBC) and various other malignancies, whereas this gene remains silenced in non-TNBC tissues and peripheral blood cells. Hypomethylation events on the X chromosome were found to be associated with aberrant CT83 activation in breast cancer, a phenomenon linked to reduced overall survival (OS) and likely involvement in the dysregulation of cell cycle signaling pathways.
Multi-omic studies, including immune profiling, have classified the microenvironment phenotypes in TNBC into 3 distinct clusters: the immune-desert cluster with low infiltration of microenvironmental cells, the innate immune-inactivated cluster with resting innate immune cells and non-immune stromal cell infiltration, and the immune-inflamed cluster with abundant infiltration of both adaptive and innate immune cells. These clusters, validated using pathological sections and external cohorts like The Cancer Genome Atlas and METABRIC, showed significant prognostic efficacy and identified potential immune escape mechanisms for each cluster. Coria-Rodriguez et al 41 used a similar approach to identify epigenomic signatures defining TNBC classes with differential therapy responses, aiming for drug repurposing.
Iqbal et al 42 studied tumor metabolic reprogramming with a multi-omic approach, identifying antagonistic roles of CBX2 and CBX7 in glucose metabolism regulation. CBX2 and CBX7 influence breast cancer metabolism through the mTOR complex 1 signaling pathway, predicting patient outcomes and drug sensitivity.
The integration of multi-omics data has significantly enhanced the prediction of tumor survival and drug response. By applying Neighborhood Component Analysis (NCA) for feature selection on datasets such as The Cancer Genome Atlas () and the Genomics of Drug Sensitivity in Cancer (GDSC), researchers have developed a neural network-based framework capable of accurately predicting survival outcomes and drug sensitivity in breast cancer patients, showing strong concordance between predicted and observed IC50 values.
Insights derived from multi-omics research are shaping novel therapeutic strategies, including immunotherapy, and are facilitating the identification of actionable targets and modulators of autophagy, such as SF3B3 and SIRT3, with potential to improve treatment outcomes in invasive breast carcinoma. The immunological and therapeutic relevance of IMMT in breast cancer has also been investigated. Multi-omics approaches have further clarified immune cell activity within breast tumors, exemplified by studies on the involvement of plasmacytoid dendritic cells in breast cancer.
These integrative strategies have unveiled key biomolecular interactions relevant to breast cancer biology, informing the development of targeted therapies. For instance, the activation of the mitochondrial protease ClpP by agents that impair essential mitochondrial functions has been elucidated in TNBC. Multi-omics analyses have also explored the dual roles of heat shock proteins as oncogenes or tumor suppressors. Marczyk et al investigated the impact of navitoclax—a BCL2 family inhibitor—on TNBC cells, identifying an 18-gene signature associated with therapeutic resistance. 43 Furthermore, DNA methylation events resulting in the silencing of HSD17B4 have emerged as predictive markers for response to HER2-targeted therapies in HER2-positive breast cancer.
Researchers have developed tools to integrate multi-omics data for breast cancer research. The Chinese Academy of Sciences implemented MOBCdb, a user-friendly database that combines genomic, transcriptomic, epigenomic, clinical, and drug response information from various breast cancer subtypes. MOBCdb offers a platform for accessing data on SNV, gene expression, microRNA expression, DNA methylation, and drug response, enhancing the visualization and analysis of multi-omics data across multiple samples.
Integrative analyses have identified recurring mutations in genes such as TP53, PIK3CA, and GATA3 in breast cancer and have revealed subtype-specific mutations like PIK3CA in luminal tumors. These multi-omics approaches have the potential to uncover new breast cancer subtypes that single datasets might miss. In addition, integrated analyses have shown the activation of signaling pathways that enhance HER2 or EGFR activity. The observed downstream phosphorylation of EGFR suggests that the HER2 signaling network activation may necessitate tailored treatment strategies for this subgroup of patients.
Integrative multi-omics analyses have revealed recurrent mutations in key genes such as TP53, PIK3CA, and GATA3 in breast cancer, while also identifying subtype-specific alterations—for example, PIK3CA mutations predominantly found in luminal tumors. These comprehensive approaches have the capacity to detect novel breast cancer subtypes that may not be discernible through single-omics data alone. Moreover, such analyses have uncovered the activation of signaling pathways that potentiate HER2 or EGFR activity. Notably, evidence of downstream EGFR phosphorylation indicates that HER2-driven signaling may be active in specific patient subgroups, warranting tailored therapeutic interventions.
In one study, integrated gene expression and proteomic data, analyzed in the context of survival outcomes among ERBB2-positive patients, identified tumors that had developed resistance to lapatinib—an inhibitor targeting the EGFR/ERBB2 signaling axis. This resistance was associated with elevated glucose metabolism, activation of the unfolded protein response, and endoplasmic reticulum (ER) stress pathways, all of which diminished the drug’s efficacy in inducing apoptosis. These findings suggest that co-targeting metabolic and signaling pathways may enhance therapeutic response in resistant tumors.
A more recent integrative study involving 122 breast cancer patients combined data on somatic mutations, mRNA and protein expression, and post-translational modifications to generate comprehensive tumor profiles. 42 This analysis delineated 4 main subtypes—basal-inclusive, HER2-inclusive, LumA-inclusive, and LumB-inclusive—that closely aligned with those defined by the PAM50 classifier. However, the integrative approach also uncovered additional biological features, such as ERBB2 amplicon status, RB alterations relevant to CDK4/6 inhibitor sensitivity, and post-translational cross-talk among proteins involved in cytoplasmic and mitochondrial metabolic pathways. In particular, acetylproteomic signatures proved valuable in differentiating luminal from basal tumors based on their distinct metabolic activities.
Lung cancer
Despite substantial research into its mutational profile and gene expression characteristics, lung adenocarcinoma (LUAD) continues to demonstrate significant intrinsic or acquired resistance to therapy. As a result, recent multi-omics initiatives have concentrated on the integration of genomic, transcriptomic, and proteomic data to uncover the molecular determinants associated with sustained responses. A large-scale multi-omics study on LUAD by the CPTAC integrated whole exome sequencing (WES), whole genome sequencing (WGS), RNA sequencing (RNA-Seq), miRNA and DNA methylation profiling, and high-resolution mass spectrometry-based proteomics, phosphoproteomics, and acetylproteomics. This integrative analysis identified 4 molecularly and clinically distinct clusters in LUAD. Cluster 1 predominantly included patients with TP53 mutations but without STK11 alterations, characterized by elevated gene expression in proximal inflammatory regions and high levels of CpG methylation. In contrast, Cluster 2 comprised TP53-wild-type tumors enriched in transcriptomic signatures associated with proximal proliferative subclusters. Ethnic stratification was observed, with Cluster 3 mainly consisting of Vietnamese patients and Cluster 4 of Chinese patients, each exhibiting unique mutational signatures. 41
Proteogenomic investigations further uncovered a novel regulatory mechanism within the KEAP1/NFE2L2 pathway involving both cis- and trans-acting elements. Interestingly, KEAP1 driver mutations did not alter transcript levels but were strongly correlated with increased NFE2L2 phosphorylation and reduced KEAP1 protein expression. This disruption of the KEAP1/NFE2L2 heterocomplex leads to the activation of antioxidant responses that promote cancer cell survival, highlighting its potential as a LUAD-specific biomarker.
Chen et al 44 applied multi-omics strategies to a cohort of early-stage, non-smoking LUAD patients in Taiwan, incorporating whole exome sequencing (WES), RNA-Seq, and proteomic profiling. Clustering analyses conducted independently across proteomic, transcriptomic, and phosphoproteomic datasets revealed that proteomic data most accurately reflected tumor stage and driver mutation patterns. Subtype 1 was characterized by advanced-stage tumors (stage > II) and a high mutation burden, including frequent TP53 mutations. Subtype 2 included stage IA/IB tumors lacking the EGFR-L858R mutation, while Subtype 3 consisted of early-stage (IA) tumors without TP53 alterations. Protein-protein interaction networks constructed using STRING elucidated the regulatory mechanisms underlying these subtypes, with extracellular matrix-associated pathways—particularly those involving MMP7, MMP11, and MMP12—significantly upregulated in Subtype 1. Immunohistochemical analysis identified MMP11 expression as a strong prognostic indicator, supporting its potential as a biomarker.
In addition, a distinct APOBEC mutational signature was identified in female patients, associated with increased expression of DNA damage response proteins and phosphosites. This finding suggests a possible link to environmental carcinogen exposure in non-smokers. 44
High-throughput technologies have revolutionized the field of biomedical research by enabling large-scale genome-wide association studies and the exploration of global transcript levels. Integrating multi-omics data in cancer research provides a systems biology approach, leveraging synergies between various molecular descriptions. However, achieving comprehensive mechanistic insights remains a significant challenge.11,16 Developing detailed genomic and transcriptomic regulatory maps that capture lung cancer complexity involves analyzing numerous gene expressions and high-dimensional genetic variants, often using multivariate regression analysis approaches. 45 Genetic regulatory connections are inherently sparse, with single variants affecting only a small fraction of gene expressions. 46
In bioinformatics research, several multi-omics methods have been developed. For example, Wang et al 47 created a precise multi-omics risk model for predicting TMB in LUAD patients by integrating gene/miRNA expression and DNA methylation data from TCGA. This model captured subtle changes in the tumor microenvironment, leading to more accurate TMB predictions. Similarly, Song et al 40 investigated the impact of Intratumor Heterogeneity on the efficacy of bispecific antibody (bsAb) immunotherapy in advanced LUAD patients. They used advanced techniques such as Digital Spatial Profiling, NGS, and the nCounter platform to analyze transcriptomic and proteomic data from over 100 Regions of Interest. These analyses aimed to unveil potential subtype-specific mechanisms, using ARACNE network analysis to scrutinize the intricate relationships between CpG sites, miRNA, and transcripts within these subtypes.
Gastric cancer
Stomach adenocarcinoma’s high prevalence and poor prognosis necessitate effective risk identification methods. Recent studies have implemented deep learning-based unsupervised approaches using multi-omics data, yielding improved prognostic performance. Multi-omics studies in gastric cancer have delineated 4 molecular subtypes: an Epstein-Barr virus (EBV)-associated subtype characterized by recurrent PIK3CA mutations; a microsatellite instability (MSI) subtype with a high mutational burden; a genomically stable subtype enriched in diffuse-type histology; and a chromosomally unstable subtype marked by aneuploidy and focal amplifications of receptor tyrosine kinases. 48
A recent proteogenomic investigation of early-onset gastric cancer validated these subtypes through integrative analysis. Phosphoproteomic data further reinforced the classification and provided insights into dysregulated signaling pathways. 45 The study employed a network propagation algorithm to integrate mutation and phosphorylation profiles, generating 2 types of network-smoothed scores. Using these scores, researchers identified 2 key cellular processes implicated in gastric cancer pathogenesis: one involving Notch and caspase signaling, and the other encompassing pathways related to MAPK, AMPK, FOXO, mTOR, and T-cell receptor signaling.
These findings highlight the potential of multi-omics approaches to identify various subtypes of gastric cancer, enhancing our understanding of patient stratification and paving the way for personalized targeted therapies.
In previous studies, deep learning-based unsupervised methods were implemented using multi-omics data and obtained better prognosis performances.43,49,50 Specifically, the autoencoder architecture in deep learning is adept at modeling complex nonlinear relationships between input and output layers, efficiently handling the high dimensionality of multi-omics data. 44 In a recent study, 2 subgroups of stomach adenocarcinoma patients were identified using an autoencoder deep learning model. The survival probabilities between these subgroups differed significantly, demonstrating the model’s superior performance compared with Principal Component Analysis and Similarity Network Fusion. This suggests that deep learning models like autoencoders better capture the nonlinear relationships in multi-omics data, enhancing prognosis predictions. The model’s robustness was validated using 10-fold cross-validation and additional independent cohorts, although high Brier scores in some datasets indicated potential errors, suggesting overall survival as a more suitable endpoint for prognosis prediction. Comparatively, autoencoders outperformed bidirectional deep neural networks in predictive accuracy, with significant differences in log-rank p values and C-index scores, suggesting that simpler models may outperform more complex ones. Differentially expressed genes, miRNAs (differentially expressed miRNAs), and differentially methylated genes between the identified subgroups highlight potential prognostic biomarkers and therapeutic targets. However, the study’s limitations include reliance on RNA-Seq, miRNA-Seq, and DNA methylation data, which may not fully capture the biological complexity, and a lack of external validation due to unavailable cohorts. Moreover, the inherent lack of interpretability in deep learning models poses a challenge to their clinical application, necessitating theoretical frameworks to elucidate model mechanics for practical use.
Other cancer types
Multi-omics strategies have also been employed in the study of various other malignancies, including pancreatic ductal adenocarcinoma (PDAC), glioblastoma, hepatocellular carcinoma (HCC), and AML. In PDAC, researchers conducted an integrative analysis of multi-omics data from 150 patients, incorporating mutational profiles, mRNA, miRNA, and lncRNA expression, DNA methylation, and proteomic data. 51 This comprehensive approach revealed significant heterogeneity in KRAS mutations and enabled the identification of distinct molecular subtypes of pancreatic cancer. Using the Similarity Network Fusion (SNF) technique for multi-omics clustering across mRNA, miRNA, and methylation datasets, 3 primary clusters were identified. These clusters were strongly associated with tumor purity and specific gene expression profiles, underscoring the importance of accounting for neoplastic cellularity in future PDAC analyses.
A pioneered study used deep learning (DL) to link multi-omics features to differential survival outcomes in HCC patients, showcasing its potential utility in enhancing HCC prognosis prediction across diverse patient populations. 52 Using RNA-seq, miRNA-seq, and methylation data from TCGA for 360 HCC patients, this DL-based model predicts patient prognosis as accurately as models that incorporate both genomic and clinical data. The model identified 2 distinct subgroups with significant survival differences and demonstrated good fitness. The more aggressive subgroup showed frequent TP53 inactivation mutations, elevated expression of stemness markers such as KRT19 and EPCAM, the tumor marker BIRC5, and activation of Wnt and Akt signaling pathways.
In Glioblastoma, multi-omics approaches have pinpointed core transcriptional factors such as CEBP and STAT3, which play roles in regulating mesenchymal transformation. 53 By integrating gene expression and phosphoproteome data, researchers have identified cellular features responsive to stress and growth factors, crucial for EGFR signaling pathway regulation and linked to patient survival.48,54,55 In addition, proteomic and metabolomic profiles have shown a unique regulatory role within a cellular network influenced by these factors. 56 A study by Dekker et al 57 using integrative multi-omics analysis on paired primary and recurrent glioblastoma tissue samples found significant differences in STMN1 (S38) phosphorylation, a key part of the ERBB4 signaling pathway.
Finally combining methylation profiles with genomic and transcriptomic data has been particularly informative in AML. A comprehensive study involving 200 adult AML patients revealed distinct gene expression and methylation patterns. 58 Significant methylation differences in CpG-sparse regions were linked to gene mutations, with AML cells harboring IDH1 and IDH2 mutations showing higher methylation levels than normal CD34+CD38− cells. Conversely, AML cells with MLL fusions or mutations in NPM1, DNMT3A, and FLT3 were associated with decreased DNA methylation, highlighting the complexity of epigenetic regulation in AML.
Table 3 summarized the multi-omics applications across cancer types.
Summary of multi-omics applications across cancer types.

Cross-cancer insights and comparative themes
Although many findings are cancer-specific, several cross-cutting themes emerge across tumor types:
Proteogenomic integration enhances predictive accuracy
In breast, lung, and gastric cancers, the integration of proteomic with genomic and transcriptomic data improves molecular subtype classification and prediction of treatment responses, such as in the activation of HER2 or dysregulation of KEAP1, NFE2L2 signaling pathways. 59
Epigenetic and metabolic reprogramming as therapeutic levers
Studies in gastric cancer and AML have identified distinct DNA methylation patterns and chromatin states associated with poor prognosis or resistance to therapy. 51 Similarly, metabolic remodeling plays a key oncogenic role in breast and hepatocellular carcinoma (HCC), highlighting potential metabolic vulnerabilities. 60
Tumor microenvironment and immune landscape stratification
Multi-omics analyses in breast, glioblastoma, and lung cancers have enabled the classification of tumor microenvironment (TME) phenotypes, such as inflamed, immune-desert, and stromal-enriched states, each increasingly linked to differential immunotherapy outcomes.61,62
Emerging insights from single-cell and spatial omics
Although not yet standard in clinical workflows, single-cell and spatial multi-omics are beginning to shed light on intratumoral heterogeneity and clonal evolution, particularly in challenging tumors such as glioblastoma and PDAC.63,64
Cross-cancer applicability of computational tools
Integration frameworks such as Similarity Network Fusion (SNF), deep learning autoencoders, and iOmicsPASS demonstrate adaptability across various cancer types. 65 However, their performance often depends on the specific combination of omics layers and the quality of the underlying data. To support clinical translation, future efforts should emphasize standardized data integration pipelines, incorporate longitudinal patient sampling, and validate omics-based biomarkers in prospective, diverse patient cohorts.
Future Direction and Conclusion
The integration of multi-omics approaches is poised to revolutionize cancer drug target discovery, offering new avenues for precision and personalized therapy. While traditional target discovery has relied on labor-intensive validation studies.66,67 high-throughput technologies now enables the comprehensive identification of therapeutic targets through multi-omics integration, enhancing both efficiency and cost-effectiveness. This has led to the discovery of critical cancer driver genes, including known targets like EGFR, ERBB2, PIK3CA, and KRAS, as well as novel candidates such as DCUN1D1 and NSD3, through large-scale analysis of like TCGA. 68 In breast cancer, multi-omics studies have revealed associations with genes such as CDK12, PAK1, PTK2, RIPK2, and TLK2, and highlighted proteomic shifts, for example EGFR overexpression following CETN3 and SKP1 loss. 69 Metabolomic insights, such as the link between glycine biosynthesis and cancer cell proliferation, further demonstrate the value of multi-omics in identifying novel therapeutic pathways. 70
Looking forward, multi-omics will play a crucial role in refining molecular subtypes, predicting drug response, and uncovering resistance mechanisms. Integrated analyses, such as those combining lncRNA, miRNA, mRNA, and methylation data, have identified chemoresistance-associated lncRNAs like HOXA-AS2 in breast cancer. 71 Similarly, proteogenomic profiling of TNBC have shown that RB protein levels, rather than genotype alone, can predict sensitivity to CDK4/6 inhibitors, except in specific resistance-conferring mutations. 29
Future research might focus on developing integrative, network-based models capable of addressing key challenges such as data heterogeneity, sparsity, and interpretability. Building hybrid frameworks that can handle both paired and unpaired omics data, as well as clinical and imaging data, will be essential. Standardized metadata schemas and integration protocols will also be critical for harmonizing datasets, prioritizing key molecular features, and improving reproducibility. 72
In summary, multi-omics integration holds immense promise for advancing cancer drug discovery. By combining robust computational methods with high-quality, unbiased datasets, researchers can achieve a deeper understanding of cancer biology, uncover shared molecular mechanisms across cancer types, and ultimately improve clinical outcomes through more effective and personalized treatments.
Footnotes
Acknowledgements
None.
Ethical Considerations and Consent to Participate
Not applicable.
Author Contributions
Conceptualization: Romina Nassini, Giandomenico Roviello
Supervision: Daniele Generali, Francesco De Logu
Investigation: Francesco De Logu
Writing – original draft: Alberto D’Angelo, Martina Catalano
Validation: Giandomenico Roviello, Daniele Generali
Writing – review and editing: Alberto D’Angelo, Martina Catalano, Romina Nassini
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.