
Abstract
BACKGROUND:
There is a current need for new markers with higher sensitivity and specificity to predict immune status and optimize immunotherapy use in colon cancer.
OBJECTIVE:
We aimed to investigate the multi-OMICs features associated with colon cancer immunity and response to immunotherapy.
METHODS:
We evaluated the association of multi-OMICs data from three colon cancer datasets (TCGA, CPTAC2, and Samstein) with antitumor immune signatures (CD8
RESULTS:
Two gene mutations (TERT and ERBB4) correlated with antitumor cytolytic activity found also correlated with improved survival in immunotherapy-treated colon cancers. Moreover, the expression of numerous genes was associated with antitumor immunity, including GBP1, GBP4, GBP5, NKG7, APOL3, IDO1, CCL5, and CXCL9. We clustered colon cancer samples into four immuno-distinct clusters based on the expression levels of 82 genes. We have also identified two proteins (PREX1 and RAD50), ten miRNAs (hsa-miR-140, 146, 150, 155, 342, 59, 342, 511, 592 and 1977), and five oncogenic pathways (CYCLIN, BCAT, CAMP, RB, NRL, EIF4E, and VEGF signaling pathways) significantly correlated with antitumor immune signatures.
CONCLUSION:
These molecular features are potential markers of tumor immune status and response to immunotherapy.
Introduction
Colon cancer is one of the most prevalent and lethal cancers worldwide [1]. Although the incidence is slightly reduced in developed countries due to the application of screening programs, colon cancer mortality remains high as patients are usually diagnosed at late stages, especially in countries with low socioeconomic status [2]. Surgery and/or chemotherapy are currently the standard treatment for colon cancer but clinical outcomes vary, although a better prognosis is generally observed in early stage compared to late stage of disease [3]. The reported immunogenic nature of some colon tumors has encouraged the development of colon cancer immune-modulating therapies. Modulation of colon cancer immune microenvironment either through enhancing tumor antigenicity (e.g. cancer vaccines), or reversing the antitumor immune response suppression (e.g. immune checkpoint therapies), have been extensively studied [4, 5]. Clinically, immune checkpoint therapies, namely anti-PD1 drugs, have produced incredible responses in treating a subset of colon cancer patients with hyper-mutated, microsatellite instable (MSI-H/MSI) tumors [5, 6, 7]. Markers of MSI status have since received increased attention in the clinical settings to guide the clinical use of PD1 blockade therapies in colon cancer [8, 9]. Microsatellite stable (MSS) cancers, on the other hand, have been invariably addressed as immunotherapy resistant cancers. Several immunogenomic studies have indicated that subgroups of MSS tumors might be responsive to immunotherapy [10, 11]. Moreover, the consensus molecular subgroups have distinct immune profiles [12]. The first subgroup (CMS1) corresponds to the previously identified MSI-H with high immune infiltration in the tumor microenvironment. The second and third subgroups (CMS2 and CMS3) are tumors with limited immune infiltration, while the fourth subgroup (CMS4) is characterized by a high stromal composition and immune-suppressing microenvironment. In each of (CMS2, CMS3, and CSM4), however, a fraction of hyper-mutated tumors was detected [12]. This overlap indicates that the previously used cancer immunotherapy markers (MSI status and tumor mutation burden), as they overlook the MSS colon tumors which might respond to immunotherapy, equally, they may also fail to predict the MSI-H subsets that responded poorly to immunotherapy. These findings highlight the need for an extensive analysis of the mechanisms regulating the tumor immune microenvironment. Understanding these mechanisms can help in identifying new and robust markers of colon cancer immunotherapy and developing effective strategies to counter immunotherapy resistance [13].
In the pursuit of more accurate or novel markers of tumor immune responses, tumor lymphocyte infiltration was verified as a reliable predictor of colon cancer clinical outcomes. The evidenced utility of immune scores in predicting patients’ survival and clinical outcome has paved the way for further research on the tumor immune microenvironment and its regulatory mechanisms [14]. The prominent advancement in OMICs technologies facilitated a comprehensive and integrative analysis of colon cancer pathogenic mechanisms [15, 16]. These technologies have recently been applied to study disease’s molecular features and their correlation with prognosis and clinical outcomes [17]. Such studies have led to the identification of important driver genes, proteins, and miRNAs in colon cancer development and progression [18, 19, 20]. In addition, several susceptibilities, diagnostic, and prognostic markers have been identified [17, 21]. Moreover, the progressive advancement in OMICs technologies has facilitated the qualitative and quantitative evaluation of tumor immune infiltration and allowed researchers to dissect key features regulating tumor immune microenvironment [13]. This will hopefully lead to the development of new immune-modulatory strategies and the identification of sensitive markers to such immunotherapy strategies [13].
In this study we applied bioinformatics analysis of three OMICs datasets of colon adenocarcinoma (COAD). Our aim was to dissect the molecular components and mechanisms that regulate tumor immune microenvironment and to highlight the clinical potentials of these as biomarkers of immunotherapy utility in colon cancer.
Methods
Datasets
We downloaded the (gene somatic mutation data, gene expression data, protein expression data, miRNA expression data, and clinical data) for the TCGA-COAD cohort (
Quantification of the immune signature enrichment levels (ISELs) in COAD samples
We analyzed the TCGA-COAD and CPTAC2-COAD samples’ immune signature enrichment levels (ISELs) using three immune signatures: CD8
ISELs associations with various OMICS features in COAD
We analyzed the TCGA-COAD and CPTAC2-COAD samples’ gene mutation data for significant associations with ISELs using the Mann-Whitney U test. We then used the Pearson correlation to identify the genes, proteins, and miRNAs with expression levels significantly correlated with ISELs. In addition, we used single-sample gene-set enrichment analysis (ssGSEA) to quantify the activity of oncogenic pathways in TCGA-COAD samples using KEGG oncogenic gene sets and identified the cancer-associated pathways (top 50 ssGSEA enriched pathways), with activities significantly associated with ISELs according to Spearman correlation test [28]. For all previous tests, we used the Benjamini Y and Hochberg false discovery rate (FDR) method [29] to adjust for multiple tests with a threshold of FDR
Gene-set enrichment analysis
We used GSEA [30] to determine the KEGG pathways that were significantly enriched for the genes having significant expression correlations with ISELs.
Survival analysis
We investigated the associations of gene mutations significantly correlated with ISELs and immunotherapy survival by comparing the overall survival (OS) between mutant and wildtype COAD samples in the three cohorts (one immunotherapy-treated cohort and two untreated cohorts). We used the Kaplan Meier survival curves to demonstrate the OS time differences with each gene and the log-rank test to evaluate the significance of OS time differences.
Cluster analysis
We also used genes with expression highly correlated (
Gene mutations associated with antitumor immune response and better immunotherapy response in COAD. A. The mutations of two genes are consistently associated with the elevated cytolytic activity in TCGA-COAD cohort (Mann-Whitney U test, FDR 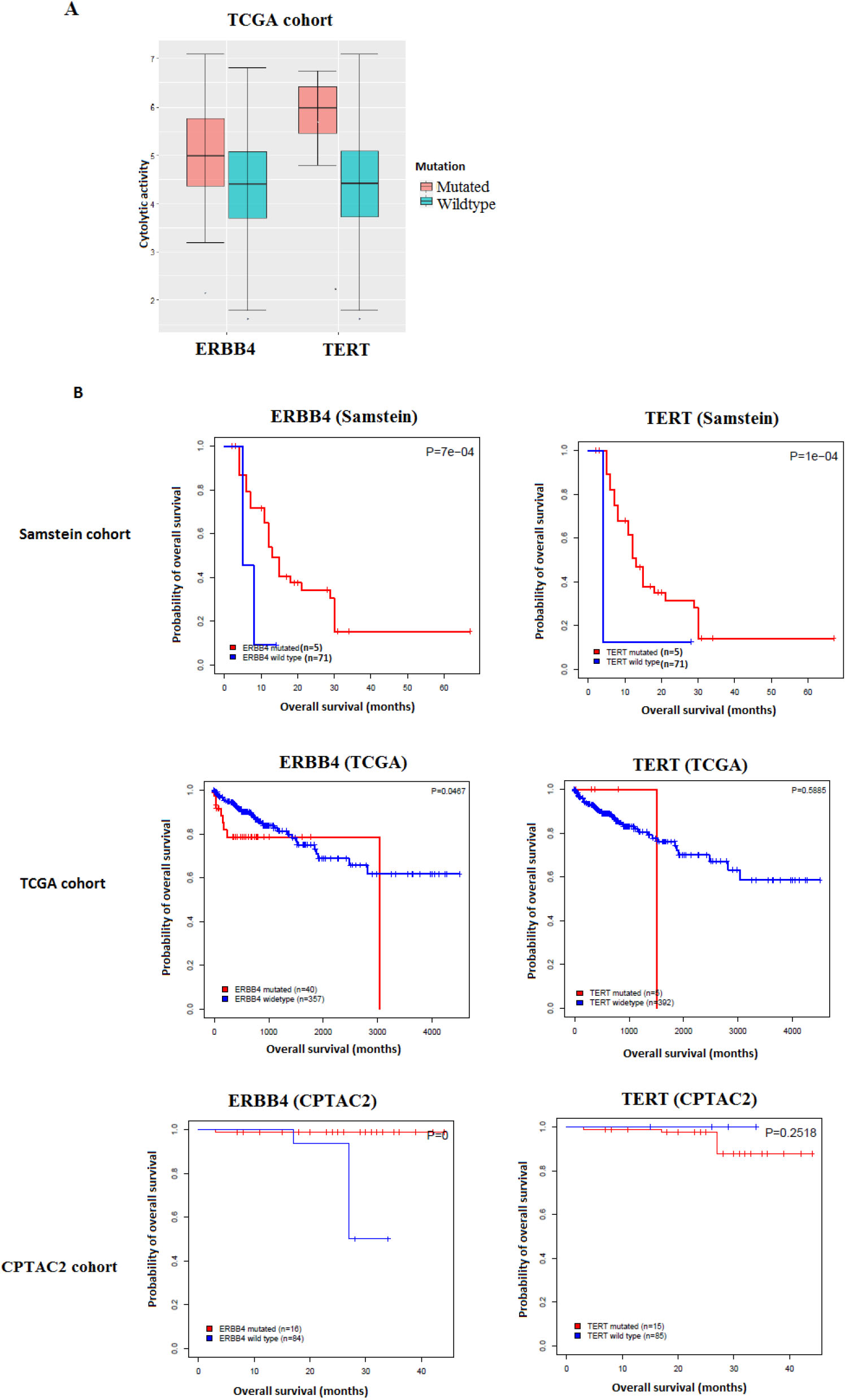
Gene mutations associated with antitumor immune response in COAD
Analyzing the TCGA-COAD cohort samples, we identified 2123 gene mutations that are significantly positively correlated with heightened CD8
We then assessed the clinical potential of the validated gene mutations by exploring their associations with OS in the immunotherapy-untreated TCGA-COAD and CPTAC2-COAD cohorts and comparing the results to those of the immunotherapy-treated (im-
Cluster analysis of TCGA-COAD and CAPTAC2-COAD datasets samples using 82 genes associated with tumor immune signatures. Cluster Heatmaps showing four clusters of TCGA-COAD and CAPTAC2-COAD samples clustered by 82 genes expression with a distinct immune profile in each cluster. Note that the immune-high cluster composes MSI-H as well as MSS and MSI-L samples, while some of the MSI-H samples clustered in classes other than the immune-high.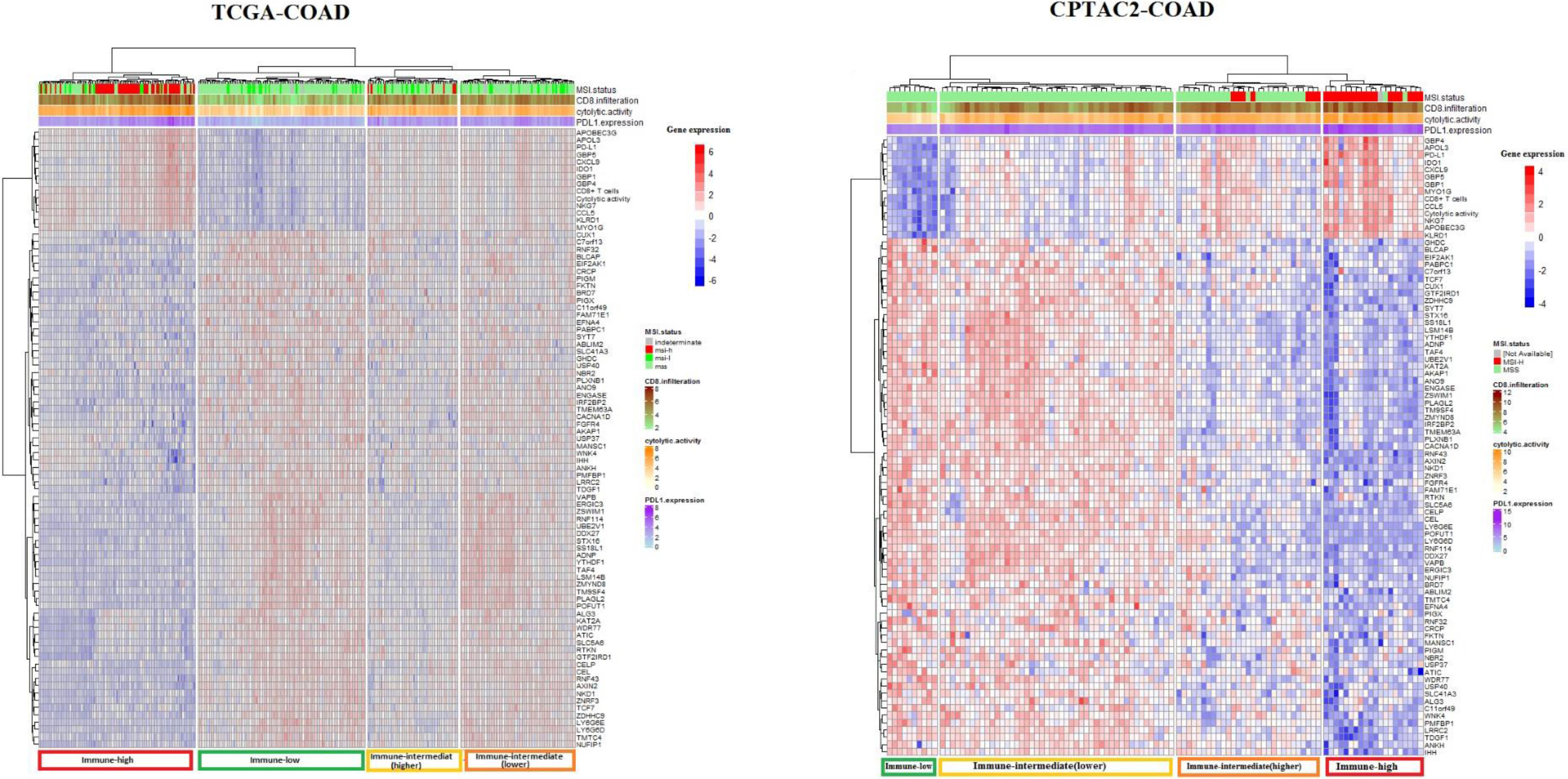
mune checkpoint inhibitors-treated) Samstein cohort. We found that the mutations of two genes (TERT and ERBB4) were correlated with elevated ICA activity and demonstrated a significant positive correlation with OS in the immunotherapy-treated Samstein cohort, while they showed no such a correlation in the immunotherapy-untreated cohorts (Fig. 1).
Protein expression levels’ association with antitumor immune response. A. Proteins expression levels associated with antitumor immune response in TCGA-COAD, B. Correlation of PREX1 protein expression levels with antitumor immune responses in TCGA-COAD and CAPTAC2 datasets, C. Correlation of RAD50 protein expression levels with antitumor immune responses in TCGA-COAD and CAPTAC2 datasets. Correlation of PREX1 and RAD50 proteins expression levels with CD8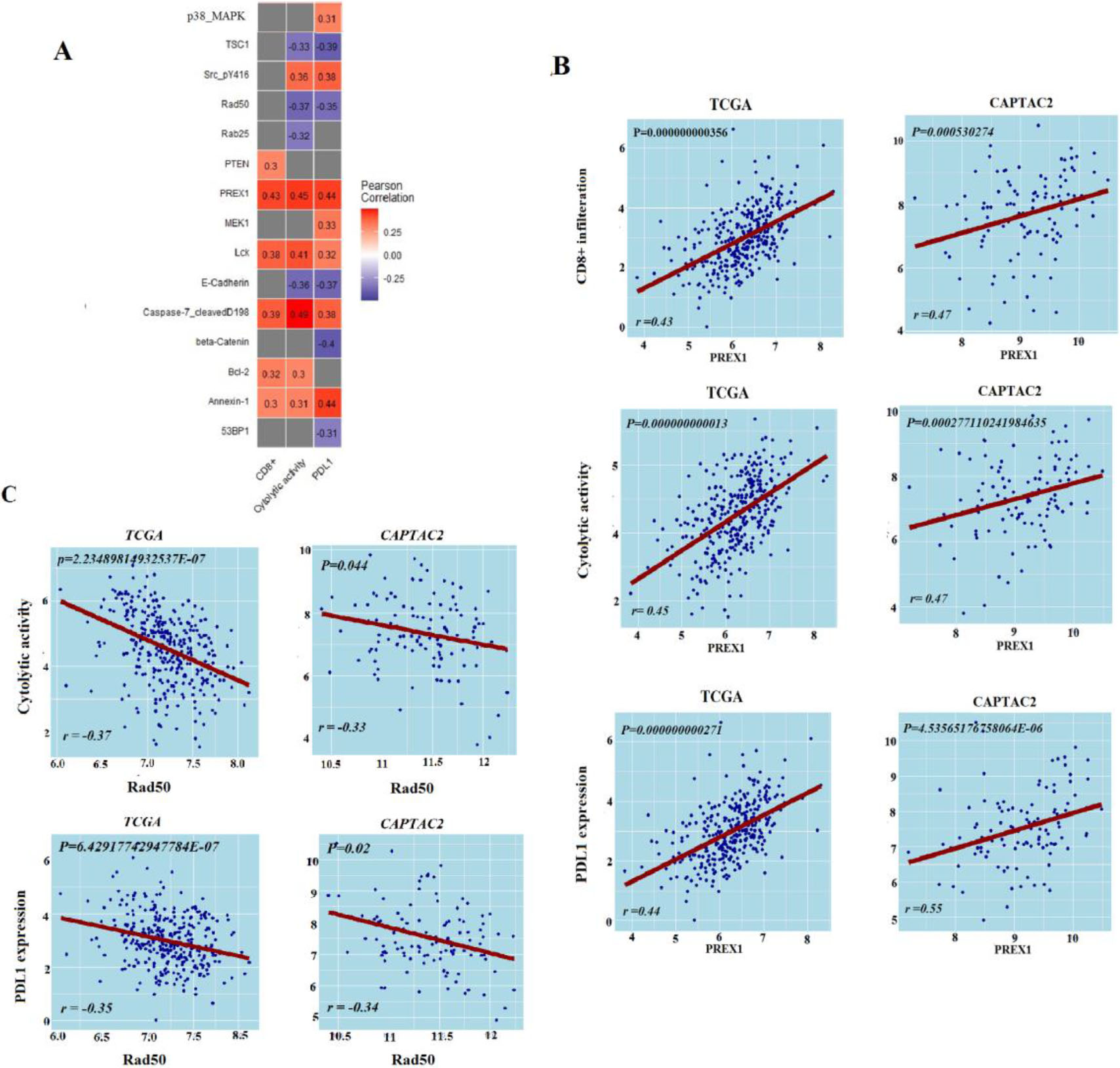
In the TCGA-COAD analysis, we found 152 genes with expression exhibiting a strong positive correlation with CD8
The TCGA-COAD gene expression data analysis produced 161 highly positively correlated genes (Pearson correlation coefficient
Similarly, the TCGA-COAD analysis reported a strong positive correlation of 111 genes’ expression levels with PD-L1 expression levels (Pearson correlation coefficient
We aimed to explore the potential of the genes we identified to have expression levels significantly correlated with ISELs in predicting tumor immune status. To do so, we filtered the genes having expression significantly correlated with the three immune signatures from the previous analysis (82 genes) (Supplementary Table S6). We used these 82 genes to perform cluster analysis using the TCGA samples for analysis and CAPTAC2 samples for validation (Fig. 2). The genes successfully clustered the samples of each dataset into four major classes with distinct immune profiles which we labeled, according to the three immune signatures’ enrichment levels in each class, “immune-high”, “immune-low”, “immune-intermediate(higher)”, and “immune-intermediate (lower)” classes. It is of note that the 82 gene clustering demonstrated greater sensitivity in identifying tumor immune phenotype compared to MSI status (Fig. 2).
Proteins with expression levels associated with antitumor immune response in COAD
From TCGA-COAD we identified five proteins (PREX1, Caspase-7_cleavedD198, Lck, Bcl-2, PTEN, and Annexin-1) with expression levels significantly positively correlated with the enrichment levels of CD8
miRNAs expression levels associated with antitumor immune response in COAD
We found that the expression of 12 miRNAs (hsa-mir-155, hsa-mir-150, hsa-mir-146b, hsa-mir-142, hsa-mir-342, hsa-mir-511-2, hsa-mir-511-1, hsa-mir-29c, hsa-mir-132, hsa-mir-140, hsa-mir-202, and hsa-mir-138-2) was significantly positively correlated with CD8
miRNAs expression levels associated with antitumor immune responses in COAD. Correlation of miRNAs expression levels with CD8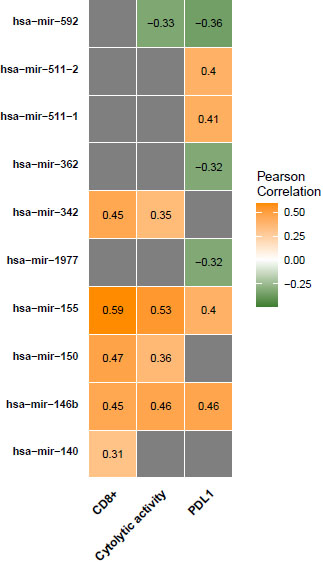
We applied ssGSEA analysis to calculate TCGA-COAD samples ssGSEA scores using oncogenic pathways gene sets. Based on these scores we investigated the correlation of the top 50 enriched oncogenic pathways in TCGA-COAD samples with ISELs. We found one pathway (CYCLIN_D1_UP.V1_DN) is positively correlated with CD8
Cancer-associated pathways whose activity is associated with PDL1 expression in TCGA CAOD. Seven cancer-associated pathways whose activity is positively associated with PDL1 expression in TCGA-COAD cohort (Spearman correlation coefficient (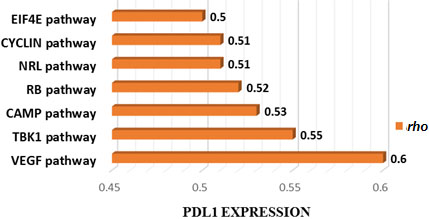
We have explored the association of gene mutations, gene expression, protein, and miRNA, from three comprehensive datasets with colon cancer anti-tumor immunity. We identified a significant number of gene mutations that are positively associated with antitumor immunity. However, the relatively limited number of the significantly correlated gene mutations replicated by the validation datasets indicated that only a limited number of these results were consistent enough to constitute a reliable marker of antitumor immune status. We filtered these validated gene mutations to further investigate association with colon cancer immunotherapy outcomes. We found two genes, TERT and ERBB4, which have mutations associated with poor survival in immunotherapy-untreated COAD patients and better survival with immunotherapy treatment. The association of these gene mutations with ICA and better prognosis when receiving immunotherapy highlights their potential as markers. The first gene, telomerase reverse transcriptase (TERT), is a known oncogene in which gain of function-mutations contribute to cell immortalization and cancer development [31]. TERT is an expressed tumor antigen in more than 80% of cancers and is recognized by both CD8
Moreover, we identified genes whose expression levels were significantly associated with antitumor immunity in COAD cohorts. We utilized the 82 genes validated by the two datasets as significantly correlated with the three antitumor immune signatures to conduct hierarchical clustering. Each dataset clustered into four clusters with differing immune profiles. The first cluster was characterized by high CD8
Protein expression data analysis, on the other hand, produced a limited number of proteins that are significantly associated with anti-tumor immunity. PREX1, previously reported as essential for TCR signaling and cytokine secretion was found positively correlated with the three antitumor immune signatures in our analysis [41]. DNA repair protein RAD50 which was already recognized as a marker of COAD poor survival and poor response to radiotherapy showed a negative correlation with the three ISELs [42]. DNA repair genes, including RAD50, have been highlighted in a previous cohort study as predictors of response to immune check-point therapy of cancer [43]. Using Bioinformatics analysis, our results validate this conclusion with RAD50. Similarly, miRNA analysis validated Six miRNAs as positively associated and four as negatively associated with antitumor immunity. Most of these miRNAs have well-described roles in processes including: fine-tuning tumor immune responses, tumor promotion, or tumor suppression [44, 45]. Hence, we suggest that the expression of the two proteins and the ten miRNAs can provide insights into the colon cancer immune status. However, additional research is necessary to verify their significance as biomarkers and/or targets for immune-modulating colon cancer therapy.
Finally, we identified few oncogenic pathways with activities significantly associated with antitumor immunity in COAD. These pathways are mainly involved in cell cycle control and tumor suppression, cell growth, cell migration, cell signaling, and immune responses [46, 47, 48, 49].
Conclusion
In conclusion, the association of the OMICs data features with COAD antitumor immune responses suggests the potential of such features in predicting the COAD patients’ prognosis and response to immunotherapy. We highlighted OMICs data features including: TERT, and ERBB4 gene mutations, an 82 gene expression signature, proteins expression of PREX1, and RAD50, and expression of 10 miRNAs, as potential markers of colon cancer immune status. Further pre-clinical and clinical studies are recommended to validate their potential as targets and/or markers of colon cancer immunotherapy.
Data availability
The supporting data (tables and figures) of this study are included within the supplementary information files.
Funding
This work was supported by the Jiangsu Province Graduate Research and Innovation Program Project (grant number KYCX21_0664 to Qiushi Feng).
Declaration of competing interest
None declared.
Author contributions
Inas Elsayed: Conception, interpretation & analysis of data, preparation of the manuscript.
Nazik Elsayed: Analysis of data.
Qiushi Feng: Analysis of data.
Kiern Sheahan: Revision for important intellectual content.
Bruce Moran: Revision for important intellectual content.
Xiaosheng Wang: Conception, revision for important intellectual content, supervision.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210222.
sj-xlsx-1-cbm-10.3233_CBM-210222.xlsx - Supplemental material
Supplemental material, sj-xlsx-1-cbm-10.3233_CBM-210222.xlsx