
Abstract
The discovery of immune checkpoint inhibitors (ICIs) has ushered a new era for immunotherapy against malignant tumors through the killing effects of cytotoxic T lymphocytes in the tumor microenvironment (TME), resulting in long-lasting tumor suppression and regression. Nevertheless, given that ICIs are highly dependent on T cells in the TME and that most tumors lack T-cell infiltration, promoting the conversion of such immunosuppressive “cold” tumors to “hot” tumors is currently a key challenge in tumor immunotherapy. Herein, we systematically outlined the mechanisms underlying the formation of the immunosuppressive TME in cold tumors, including the role of immunosuppressive cells, impaired antigen presentation, transforming growth factor-β, STAT3 signaling, adenosine, and interferon-γ signaling. Moreover, therapeutic strategies for promoting cold tumors to hot tumors with adequate T-cell infiltration were also discussed. Finally, the prospects of therapeutic tools such as oncolytic viruses, nanoparticles, and photothermal therapy in restoring immune activity in cold tumors were thoroughly reviewed.
Introduction
Immune checkpoint inhibitors (ICIs) have demonstrated remarkable benefits in patients with various advanced malignancies, ushering in a new age of cancer immunotherapy. Among the numerous immunomodulators, the most advanced is the development of drugs that target programmed death protein 1 (PD-1), an immune checkpoint receptor located on T cells. Programmed death-ligand 1 (PD-L1) can bind to PD-1 to cause T-cell depletion and immunosuppression. 1 Thus, inhibition of the PD1/PD-L1 pathway has a major impact on the restoration of antitumor immune effects. Although ICIs are extremely effective in the treatment of advanced melanoma, metastatic lung cancer, and other diseases, patient outcomes remain poor.2,3
Tumors can be classified as immunoinflammatory, immune-excluded, or immune-desert based on the infiltrative distribution of cytotoxic immune cells in the tumor microenvironment (TME). Researchers often refer to immunoinflammatory tumors as hot tumors, which are typically accompanied by high T-cell infiltration, PD-L1 expression, and tumor mutational load (TMB), thereby enabling tumor cells to respond to ICI therapies. Immune rejection of tumors is often associated with cytotoxic T lymphocytes (CTL) being blocked at the surface of the tumor and unable to infiltrate the tumor to exert their killing effect. Immune-desert tumors, also referred to as cold tumors, are characterized by a TME with a low number of CD8+ T cells. Meanwhile, tumor-associated macrophages (TAMs), T/B regulatory cells (T/Bregs), myeloid-derived suppressor cells (MDSCs), and a hypoxic microenvironment were found to create a specific immunosuppressive TME in “cold tumors”.4,5 At the same time, cold tumors are virtually unresponsive to ICIs owing to their low mutational load, antigen-presenting capacity, and PD-L1 expression. 6
Therefore, T-cell infiltration into the TME and their immunocidal effects are essential for the progressive conversion of cold tumors to hot tumors. Herein, we summarize the mechanisms by which cold tumors are highly immunosuppressed by the TME, the results of decades of attempts to enhance the immunosuppressed state of the TME, and the prospects of combining ICIs with hot and cold conversion therapies against tumors.
Mechanisms of TME-Induced Immunosuppression in Cold Tumors
The formation of cold tumors can be attributed to numerous complex immune evasion mechanisms, of which the complexity of the TME plays a vital role. Immunosuppressive cells, transforming growth factor-β (TGF-β), STAT3 signaling, adenosine, and the formation of physical barriers and intricate vascular networks within the tumor all play an essential role in the development of an immunosuppressive environment in the TME. Their presence continues to drive progress in the immunosuppressive microenvironment.
Immunosuppressive cells in the TME promote cold tumor progression
The TME is a dynamic network structure with inherent complexity during cancer progression or in response to different therapeutic modifications. 7 The presence of immunosuppressive cells (primarily MDSCs, Tregs, and TAMs) in tumors constitutes a vital source of tumor immunosuppression.8,9 The role of immunosuppressive cells in cold tumor formation is illustrated in Figure 1. Notably, MDSCs are immunosuppressive cells that promote tumor growth in the TME. 10 They can suppress the immune response through multiple mechanisms. On the one hand, MDSCs can downregulate the TCR-associated ζ-chain, leading to the failure of CD4+ and CD8+ T-cell activation signaling and a severe paucity of T cells in the TME. On the other hand, MDSCs express high levels of arginase 1 (ARG1), 11 which catabolizes the amino acid arginine that is essential for T-cell activation in the environment, and generates large amounts of nitric oxide (NO) in the TME, thereby inhibiting CD4+ and CD8+ T-cell activation.11,12 Furthermore, MDSCs mediate immunosuppressive effects via TGF-β, which induces Treg accumulation and regulates MDSCs. Tregs can exert their potent immunosuppressive functions through several mechanisms. In the TME, they generate large amounts of cytokines such as interleukin (IL)-10, IL-35, and TGF-β, which inhibit the antitumor immune activity of cytotoxic T cells. 13 By expressing CTAL-4 to compete for CD80/CD86 on antigen-presenting cells (APCs), Tregs can further influence the antigen-presenting effect of immune cells and thus exert a potent immunosuppressive effect. Compared with active Tregs, apoptotic Tregs can more effectively inhibit CTL activation. In the TME, reactive oxygen species (ROS) target mitochondria to induce apoptosis, whereas apoptotic Tregs achieve a substantial immunosuppressive effect through oxidative stress–related mechanisms, leading to a highly immunosuppressive TME. In a tumor-bearing mouse model, the researchers observed that apoptotic Tregs were highly resistant to the therapeutic effects of PD-L1 inhibitors. 14 Therefore, given the immunosuppressive effects of apoptotic Tregs, conventional therapies for the depletion and functional regulation of Tregs may be ineffective, and researchers are seeking to develop drugs to prevent Tregs from entering the TME to create a suitable TME for immunotherapy. 15 Tumor-associated macrophages are pivotal cells driving the formation of an immunosuppressive TME and achieve their suppressive function on T cells by secreting the cytokines TGF-β and IL-10. 16 - 18 Moreover, they can stimulate the release of the chemokines CCL5, CCL20, and CCL22, which contribute to Treg recruitment into the TME to exert their immunosuppressive effects.19,20 More importantly, TAMs can synthesize matrix metalloproteinases (MMP2 and MMP9), IL-6, urokinase-type plasminogen activator (u-pA), and tissue-type plasminogen activator (t-pA) to degrade the extracellular matrix (ECM), thus facilitating tumor growth. 21
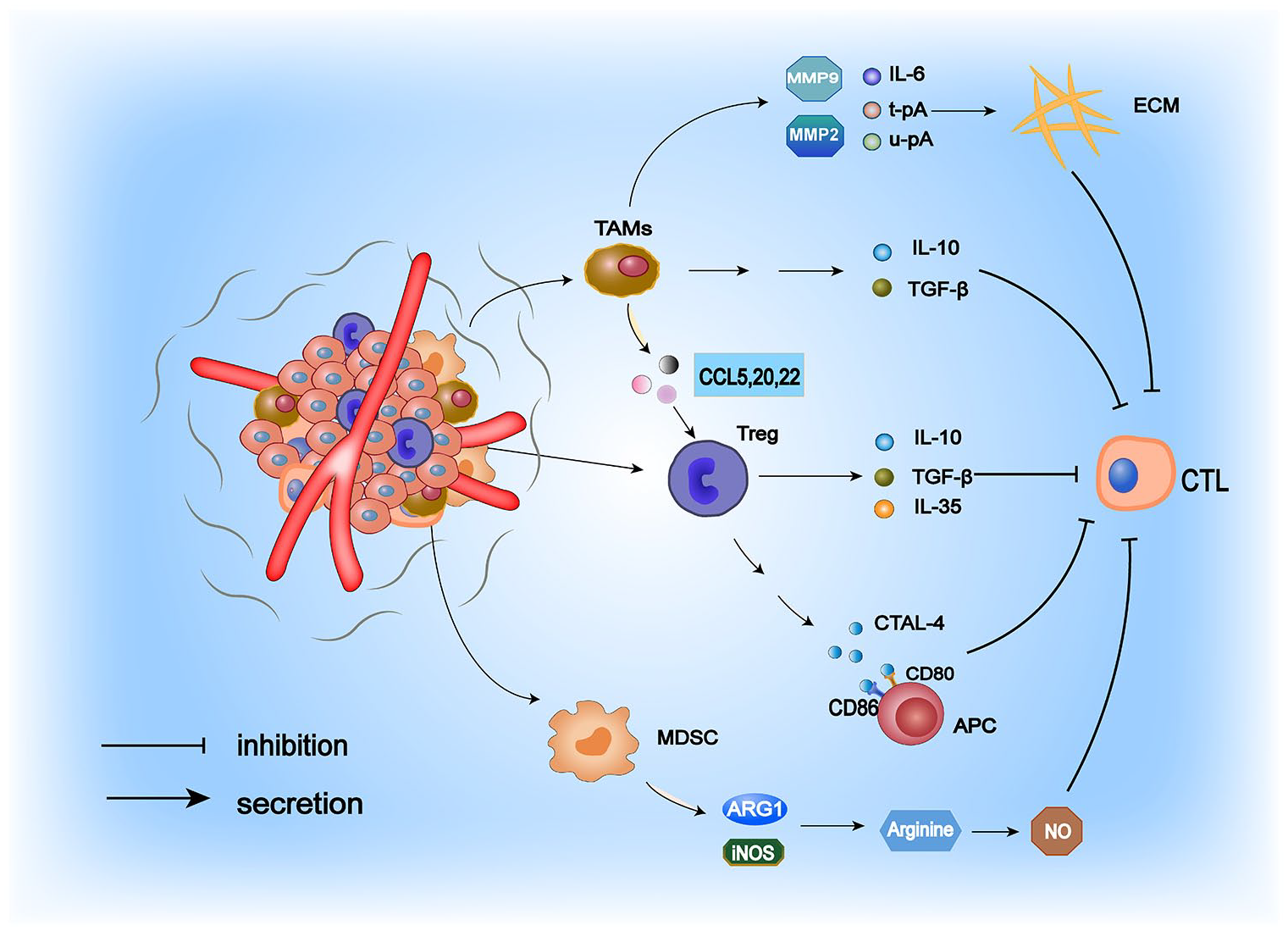
Mechanisms by which the central immunosuppressive cells in the TME function. TAMs, Tregs, and MDSCs create an immunosuppressive TME by recruiting various factors, which in turn produce a robust inhibitory effect on CTL.
TGF-β and STAT3 signaling promote the development of an immunosuppressive TME
Transforming growth factor-β plays a vital role in the development of the immunosuppressive TME. (1) TGF-β secretion from cancer cells and mesenchymal fibroblasts in the TME induces dendritic cells (DCs) to downregulate cytokines that promote inflammatory responses as well as their antigen-presenting abilities, resulting in reduced T-cell expression.22,23 (2) TGF-β triggers the synthesis of indoleamine 2,3-dioxygenase (IDO),24,25 which in turn lowers tryptophan levels in tumors to suppress the proliferative capacity of T cells. 26 (3) In the presence of TGF-β, cancer cells undergo epithelial-mesenchymal transition (EMT), which promotes their migratory and invasive properties and triggers the conversion of CD4+ T cells to in situ suppressive Tregs. 27 (4) Transforming growth factor-β induces the upregulation of vascular endothelial growth factor, affecting aberrant blood vessel formation within the TME. 28 The abnormally vascularized TME leads to immunosuppression and a hypoxic state. At the same time, it increases the interstitial pressure and results in inadequate perfusion within the tumor, making it difficult for T cells to penetrate the TME.29,30 (5) Transforming growth factor-β signaling also stimulates Smads and the transcription factor ATF1 to inhibit granzyme B and interferon-γ, which suppress the immune effects of CD8+ T cells and promote CD8+ T-cell lysis. 31 All of the above collectively contribute to the development of a cold tumor. The hypoxic tumor environment can inhibit CD8+ T-cell infiltration and function by activating STAT3 signaling in target cells, promoting tumor cell survival, proliferation, and the recruitment of TAMs and Tregs into the TME, and moderately facilitates communication between the tumor and various immune cell subsets. 32 The mechanism by which hypoxic TME leads to cold tumor formation is delineated in Figure 2. In addition, the activation of the STAT3 signaling pathway leads to the inhibition of dendritic cell maturation and has an antitumor effect by inhibiting CTL and natural killer (NK) cells, thereby preventing tumor cold-to-hot transition. 33

Atypical tumor vascular growth leads to hypoxic TME formation, which is an important mechanism underlying tumor immune evasion. This attracts large numbers of MDSCs and TAMs, and tumor cells can secrete high levels of the cytokine CCL28 under hypoxic conditions, thereby recruiting large numbers of Tregs (immunosuppressive cells) into the TME. Tregs can survive hypoxic conditions and, in response, regulate CD73 and CD39, both of which convert ADP and ATP into adenosine. Adenosine inhibits the proliferation and expansion of T cells and the secretion of effector cytokines by binding to the adenosine A2A receptor on T cells, depriving them of their aggressiveness against tumor cells 34 . Meanwhile, the presence of adenosine contributes to the increase in the number of Tregs. In addition, it promotes their immunosuppressive effects, with positive feedback between the two continuously driving the immunosuppressive environment of cold tumors. The presence of high adenosine levels in the TME significantly advances the progression of tumor immune escape. 35
Formation of a physical tumor barrier induces immunosuppression in the TME
Cancer-associated fibroblasts (CAFs) are thought to originate from fibroblasts activated by inflammatory and tumor signals. The activation of TGF-β/Smad, ERK/MAPK, and AKT-mTOR signaling in the TME promotes their activation and differentiation. Cancer-associated fibroblasts generate ECM and continuously converge to form a dense physical barrier outside the tumor, preventing the infiltration of antitumor drugs and T cells. 36 They also secrete other cytokines, chemokines, and growth factors that suppress the activity of CD8+ T cells and recruit immunosuppressive cells, enabling tumor cells to evade immune surveillance.37,38
There is a dynamic balance between provascular and antivascular signaling in healthy tissues that maintain the standard vascular architecture. In contrast, hypoxia in the tumor environment leads to tumor microvascular disturbances,29,30 affecting the delivery of chemotherapeutic drugs and nutrients and inhibiting immune effector T-cell function and antitumor immunity. 39 During tumor development, TGF-β has been proven to induce a proangiogenic environment and stimulate tumor angiogenesis both in vitro and in vivo. The tumor vasculature acts as an essential barrier to T cells by deregulating the adhesion molecules required for T-cell extravasation. 40 This results in fewer T cells entering the TME, thereby exacerbating the immunosuppressive microenvironment of the “cold” tumor.
Weak antigenicity of the tumor and impaired antigen presentation lead to immune escape
The human immune system predominantly relies on antigens on the surface of tumors to specifically recognize tumor cells, whereas lesser antigenic tumor cells are harder to recognize. Tumor-associated antigens (TAAs) and cancer germline antigens (CGAs), 2 common tumor antigens, are expressed in both tumor and healthy human tissues (eg, testis and fetal ovary), 41 - 43 raising the possibility that treatments targeting TAAs and CGAs may lead to severe toxicity attributed to immune activation in healthy tissues. 44 Furthermore, the involvement of central and peripheral immune tolerance mechanisms renders treatments targeting TAA and CGA ineffective. 45
During antigen presentation, dendritic cells play an instrumental role in activating the cellular immune response by taking up, processing, and presenting antigens to effectively destroy tumor cells. 46 However, recent studies have established that the production of ROS impairs the function of DCs in the TME and activates the endoplasmic reticulum (ER) stress response factor XBP1, which in turn promotes the synthesis of triglycerides and lipids in dendritic cells, eventually leading to lower antigen-presentation ability and inducing immunosuppression in the TME. 47
Tumor cells can escape immune surveillance by downregulating major histocompatibility complex (MHC). MHC-I deficiency prevents the immune system from detecting tumor cells, 48 leaving cytotoxic T cells with a limited role. In a low glycemic and hypoxic environment, the expression level of MHC-I on the surface of tumor cells is downregulated. As a result, the hypoxic environment in most TMEs contributes to the evasion of tumor cells from the immune system. 49 Mutations in the MHC-I gene in malignant tumors may completely abolish its peptide loading or alter the peptide pool that binds to MHC-I, leaving MHC-I ineffective at recognizing CD8+ T cells. Moreover, decreased levels of interferon-γ (IFN-γ) in cancer cells can also lead to the downregulation of MHC-I.50,51 NLRC5 is a crucial modulator of IFN-γ that upregulates MHC-I expression; 48 and lower NLRC5 activity is closely correlated with MHC-I downregulation and can result in immune evasion. 52 Mutations in oncogenes are also critical for the downregulation of MHC-I. Besides, overexpression of n-myc and c-myc may lead to the silencing of MHC-I expression by affecting the NF-κB pathway. 53 - 55 It has been reported that the latter can downregulate MHC-I in gliomas through the Wnt/B-catenin pathway, thereby resulting in immunosuppression. 54
Inactivation of the IFN-γ signaling pathway induces tumor immunosuppression
The release of IFN-γ into the TME to kill tumor cells is an essential approach by which T cells kill tumor cells. The biological effects of IFN-γ are mainly mediated through the JAK/STAT (signal transducer and stimulator of transcription) pathway. When IFN-γ binds to the tumor surface receptor (IFNγR), IFN-γ is phosphorylated, resulting in the activation of the downstream signaling components JAK1 and JAK2. The intracellular region of the activated JAK-phosphorylated receptor creates a binding site for STAT1, which subsequently activates IRF1 to upregulate PD-L1 levels and inhibit T-cell activity. The use of a PD-L1 inhibitor subsequently relieves T-cell suppression and activates the antitumor effect of the immune system. However, tumor development is often accompanied by mutations in JAK1 and JAK2, which affects the JAK/STAT pathway and the level of PD-1/PD-L1 in vivo, resulting in nonresponse of the immune system. 56 In contrast, the downstream pathway of JAK1/2 governs the expression of chemokines such as CXCL9, CXCL10, and CXCL11, which have a powerful recruitment effect on T cells. In addition, mutations in upstream JAK1/JAK2 downregulate the expression of downstream chemokines, which may also contribute to poor T-cell infiltration in cold tumors. 57
Mitochondrial hijacking assists in immune evasion in the TME
Cancer cells can hijack the mitochondria of immune cells through nanoscale tubular structures, allowing functionally defective and obsolete mitochondria in cancer cells to be substituted by new functional mitochondria for maximum efficacy. 58 Mitochondria in the normal physiological state in cancer cells enable sustained glycolysis in the TME by stabilizing hypoxia-inducible factor 1 (HIF-1α), which supplies cancer cells with lipids, nucleotides, and proteins required for anabolic metabolism. Meanwhile, pyruvate, an intermediate product of aerobic glycolysis, can neutralize intracellular ROS. Mitochondria, in their natural state in cancer cells, may suppress the immune system through the following mechanisms. (1) Sustained glycolysis in mitochondria leads to TME acidosis through efflux to the extracellular environment, while the acidic TME disables immune cells. 59 (2) Hijacking of mitochondria from T cells allows depletion of the energy source of immune cells while potentially overexpressing PD-1 and thus inhibiting antitumor efficacy.60,61 (3) Mitochondria participate in various immunosuppressive activities regulated by HIF-1α through its stabilization, such as (a) regulation of glucose transporter protein (GLUT-1) expression, which causes glucose to go beyond the range of immune cells, leading to depletion of energy in immune cells and failure to exert antitumor effects; 62 (b) downregulation of MHC-I in the hypoxic TME precipitates the downregulation of antigen presentation; 63 (c) modulation of the release of chemokines from cancer cells; and (d) HIF-1α suppresses the immune system by directly binding to its promoter to upregulate the gene expression of IL-10 and TGF-β. 64 Conversely, as mitochondria within T cells are hijacked by cancer cells, the presence of impaired mitochondria in T cells brings about rapid depletion of intracellular adenosine triphosphate (c-ATP), which is essential for T-cell activation and migration, resulting in PD-1 treatment having only short-term effects. 61
Converting Cold Tumors Into Hot Tumors (The Specific Method Is Detailed in Table 1)
Current approaches to driving T-cell proliferation in the TME.

Abbreviations: MDSC, myeloid-derived suppressor cell; TGF-β, transforming growth factor-β; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
T-cell initiation by oncolytic viral therapy
Oncolytic viruses are natural or recombinant viruses designed to target and kill tumor cells. The viruses cause cancer cells to die at the end of their replication cycle via lysis or activating an antitumor immune response, avoiding damage to healthy cells. At the same time, viral infection stimulates the generation of antitumor immune responses and promotes the development of antitumor immunity. 65 Antigen-presenting cells such as dendritic cells can capture tumor-associated and neoplastic antigens released by virus-mediated lysis of tumor cells, stimulating a broad-spectrum antitumor response in immune cells. Moreover, the oncolytic virus can promote immunogenic cell death to initiate the release of danger-associated molecular patterns (DAMPs). The DAMPs and pathogen-related molecular patterns (PAMPs) are then recognized by DC pattern recognition receptors, which promote the recruitment of additional T cells in the TME to exert antitumor immune effects.74,75 Meanwhile, the oncolytic virus attacks tumor stromal cells to disrupt the complex tumor architecture. 76 On the one hand, the oncolytic virus can break through the physical barriers of necrosis, hypoxia, and high interstitial pressure to penetrate and act within the tumor. On the other hand, the oncolytic virus perturbs the intricate mesenchymal stroma of the tumor and promotes T-cell infiltration to exert their antitumor effects. More importantly, the oncolytic virus stimulates the production of CXCL9 and CXCL10 to upregulate the expression of selectins and integrins, promoting T-cell infiltration into the tumor and exerting an antitumor immune effect. 77
Improving tumor antigen presentation to enhance the immunosuppressive TME
Targeting tumor neoantigens to improve tumor antigen-presentation disorders
Tumor neoantigens play a decisive role in tumor-specific T-cell-mediated antitumor immune responses. 66 Neoantigens are nonautologous proteins with unique specificity produced by nonsynonymous mutations in the genome of tumor cells. 78 They have greater immunogenicity and higher affinity for the MHC and are unaffected by central immune tolerance. 79 Tumor mutational load is a measure of the number of mutations in cancer cells, and mutations in tumor cells can generate neoantigens that are presented to T cells by MHC proteins, thereby better priming the immune system.80,81 Theoretically, a higher TMB allows for the production of more neoantigens, leading to an increased likelihood of T-cell recognition and contributing to an improved response to ICIs. 82 However, not all tumor patients with high TMB respond well to immunotherapy. Immunotherapy is primarily used for the treatment of cancers with a high mutational burden, but there is also the possibility of identifying neoantigen-targeted immune cell responses in tumors with intermediate/low mutational burden. 83 Thus, there is an urgent need to develop vaccines against neoantigens to convert immune “cold” tumors to “hot” ones. Nevertheless, the development of novel antigens is complex. Tumor growth is an evolutionary process, and the genomes of tumor cells are constantly mutating as they grow. Vaccines developed for the genome of a single tumor sample often have little success when the neoantigen is a subclone of a mutated tumor gene. 79 Furthermore, the variation in neoantigens produced by tumors in different patients makes treatment against neoantigens extremely challenging.84,85
Upregulation of MHC-I restores the immune system’s ability to recognize tumors
Downregulation of MHC-I is one of the critical mechanisms underlying tumor immune escape, resulting in poor recognition of the immune system and tumors and thus the inability of the body to function as an antitumor immune agent. Therefore, the development of drugs that restore MHC-I expression may be an ideal approach to reestablish the immune effect of CTL in cold tumors. (1) Targeting NLRC5 to restore MHC-I levels; when NLRC5 is upregulated, it combines with atf1/CREB, RFX, and NFY complexes in the nucleus to form MHC enhancers, which in turn bind to the SXY module of the MHC-I promoter to further boost the transcriptional activation of MHC-I.86,87 In addition to MHC class I genes, NLRC5 induces the expression of β2M, TAP1, and LMP2, which are essential components for MHC class I antigen presentation, thereby restoring tumor immunogenicity. 88 (2) Natural killer cell therapy restores MHC-I levels; NK cells identify and remove cells with downregulated or absent MHC-I expression by releasing cytotoxic particles or binding TNF receptor superfamily death receptors on target cells to ligands expressed by NK cells. 48 For example, in patients with periampullary adenocarcinoma, pancreatic adenocarcinoma, and non–small cell lung cancer, significant infiltration of NK cells often correlates with a better outcome.89,90 (3) Increased expression of IFN-γ to restore tumor MHC-I levels: interferon activates the JAK/STAT pathway, which phosphorylates and dimerizes the STAT that is present in an inactive form in the envelope and acquires a migratory invasive phenotype, which then upregulates the expression of MHC-I. 48 Meanwhile, IFN-γ can also induce NLRC5 expression to further form CITA enhancers and induce an increase in MHC-I expression. 86 (4) Inhibition of autophagy restores MHC-I levels: although downregulation of MHC-I expression is frequently observed in most malignancies (eg, pancreatic ductal adenocarcinoma [PDAC]), 86 gene mutations leading to MHC class I deficiency have rarely been reported.91,92 Previous studies have established that NBR1-mediated selective autophagy of MHC-I is one of the mechanisms that promote immune evasion in PDAC cells and that inhibition of autophagy in PDAC alters surface MHC-I levels and allows for improved antigen presentation. 93 Furthermore, enabling CD8+ T cells to recognize tumor antigens more efficiently further promotes their activation to enhance antitumor responses.
Activation of dendritic cells by CD40 agonists to improve impaired tumor antigen presentation
CD40 is a cell surface member of the TNF receptor superfamily and is abundantly expressed on immune cells such as DCs, B cells, and macrophages, where it plays a permissive role in the activation of antitumor T cells by DCs. One of the critical steps in activating CD8+ T cells by antigen-presenting cells is accomplished by receiving signals from CD40L on CD4+ T cells, which subsequently stimulates CD40 on the surface of antigen-presenting cells. 67 Moreover, CD40 enhances the synthesis of IFN-γ and IL-12 and the immunosuppressive microenvironment. 94 Prior studies have reported that CD40-activated macrophages can also exert a destructive effect on the tumor mesenchyme, thus partially facilitating the delivery of chemotherapeutic drugs to the internal tumor environment. 95 The use of activated CD40 antibodies has been observed to contribute to the infiltration of large numbers of CTL cells in PDAC mice and lead to tumor regression. 96 In another study, the combination of gemcitabine and CD40 agonists also demonstrated tumor cell clearance in a phase I trial in patients with PDAC, 97 implying that CD40 agonists combined with antitumor agents may assist in enhancing the therapeutic effect on cold tumors.
Using cancer vaccines to enhance antigen presentation
Cancer vaccines are predominantly used to induce specific cellular and humoral immune responses by activating the patient’s immune system and using tumor cells or tumor antigens to trigger the proliferation of tumor-specific T cells for tumor clearance.98,99 They are based on tumor-associated antigens and are chiefly classified as cellular vaccines (tumor and dendritic cell vaccines), genetic vaccines, and peptide vaccines. The key to cancer vaccine development is to identify the optimal vaccination antigen, which is selected from tumor cells and presented via MHC-specific peptides, DNA-encoded proteins, and recombinant viral or bacterial vectors expressed in DSC. 100 GVAX is one of the vaccines developed to treat pancreatic cancer but failed to improve survival in phase IIb/III trials in patients with metastatic PDAC. However, GVAX vaccination showed increased tertiary lymphocyte formation and T-lymphocyte infiltration and promoted the upregulation of PD-1/PD-L1, 101 suggesting that vaccination may sensitize tumors to immune checkpoint blockade therapy. 7 Collectively, vaccination has the potential to effectively initiate or amplify antitumor T-cell responses.
Modulation of the intestinal flora for improved antigen presentation
The metabolism of the intestinal flora is one of the most critical factors in maintaining the human immune system, and there are approximately 1000 microorganisms present in the human gut that can reshape the TME by modulating the host immune system, thus contributing to increased activity against PD-1/PD-L1 or CTAL-4 ICIs.102,103 Current research on gut flora and ICIs is presented in Table 2. Neutrophil accumulation plays a pivotal role in driving tumor progression, with tumor cells recruiting neutrophils via Toll-like receptor 4 (TLR4) signaling, which triggers metastasis to endothelial cells by releasing tumor necrosis factor. In contrast, the intestinal flora has been found to contribute to neutrophil reduction, signaling that it may have tremendous potential in inhibiting cancer progression. 104 Bifidobacteria induce DCs to promote the differentiation of primary T cells to CD25+ FOXP3+ Treg cells, resulting in an immunosuppressive state. 105 The accumulation of Bifidobacterium in the TME significantly improves the effect of anti-CD47 immunotherapy, while CD47 plays an active role in aiding tumor cells to evade immune system surveillance. Moreover, blocking CD47 can restore the immune system’s ability to monitor tumor cells by sending a “don’t eat me” signal to the immune system. More importantly, Bifidobacterium enhances the effect of anti-CD47 antibody therapy through DC-specific type I interferon and the activation of the host cGAS-STING pathway mediated by mitochondrial DNA in DCs. 106 - 108 In addition, the ability of Bifidobacterium to promote DC maturation and lymphocyte activation results in enhanced CD8+ T-cell priming and CD8+ T-cell aggregation in the TME, indicating that Bifidobacterium also plays a key role in lowering the activation threshold of DCs, allowing for lower antigen concentrations to prime more T cells, and driving T-cell proliferation with superior antitumor effects. 102 Some bacterial metabolites, such as inosine and single-chain fatty acids, are also hypothesized to participate in TME remodeling to influence cancer immunotherapy.68,109 According to a study conducted by Sivan et al, 110 mice receiving Bifidobacterium were significantly more responsive to antitumor drug treatment. In addition, a 16sRNA classification survey of pretreatment and posttreatment stool samples from patients with advanced gastrointestinal cancers treated with anti-PD-1/PD-L1 therapy demonstrated that the higher the ratio of Prevotella to Bacteroides in the body, the more effective the PD-1/PD-L1 inhibitors. 111 Research into the interaction between gut flora and tumor immunotherapy is still in its infancy, and the mechanisms involved still need to be further explored in the context of extensive in vivo, ex vivo, and clinical data. However, the results of established mouse models and current clinical data suggest that altering the gut micro-ecosystem to improve the highly immunosuppressive TME of cold tumors is a promising approach. 112
Studies combining immune checkpoint inhibitors and gut microbiota.

Data collected from https://clinicaltrials.gov/
Abbreviations: PD-1, programmed death protein 1; PD-L1, programmed death-ligand 1.
Enhancing T-cell infiltration to improve immunosuppression in the TME of cold tumors
Targeting MDSCs to restore the antitumor activity of the immune system
Myeloid-derived suppressor cells play a potent and widespread role as immunosuppressive cells.4,113 Currently, MDSC expression in the TME is primarily lowered via depletion of MDSCs, inhibition of MDSC recruitment to the tumor, and promotion of MDSC differentiation. 11 In the strategy of depleting MDSCs, targeting the TNF-related apoptosis-inducing ligand receptors (TRAIL-Rs) may be a practical approach. Besides, targeting TRAIL-Rs may limit the activity of MDSCs through ER stress. 114 Moreover, miR-155 and miR-21 overexpression was found to promote the expansion of MDSCs in monocytes and granulocytes. Taken together, the regulation of miR-155/miR-21 may contribute to the depletion of MDSCs. 115 Some chemotherapeutic agents such as gemcitabine, 5-fluorouracil (5-FU), and paclitaxel have been reported to eliminate MDSCs to some extent in mouse tumor models when used at low doses, 116 - 119 with significant antitumor effects. Sildenafil, a phosphodiesterase (PDE-5) inhibitor, suppressed the activity of nitric oxide synthase and ARG1, thereby exerting an inhibitory effect over the function of MDSCs and significantly increasing the survival rate of mice.120,121 In the strategy of inhibiting the recruitment of MDSCs to tumors, CC chemokine ligand 2 (CCL2) and its receptor (CCR2) are predominant drivers of migration of MDSCs to the TME, and the use of CCL2-neutralizing antibodies and CCR2 antagonists has been demonstrated to significantly lower the expression of MDSCs in preclinical tumor models. 122 In addition, the development of a STAT3 inhibitor (AZD9150) and the use of all-trans retinoic acid have shown encouraging results in inducing MDSC differentiation into immunogenic dendritic cells or macrophages.123,124
Restoration of antitumor immune activity by TGF-β inhibitors
Activation of TGF-β plays an instrumental role in driving endothelial cell transformation and angiogenesis to promote tumor progression and conferring resistance to chemotherapy. 125 The development of therapies targeting TGF-β has been validated as one of the key methods by which T-cell infiltration can be restored. 126 - 128 Galunisertib is a small-molecule inhibitor of TGF-β that blocks TGF-β signaling through TGF-β receptor I signaling. 129 Its combination with gemcitabine improved the overall survival of patients with unresectable pancreatic cancer, while the toxic effects of the drug on patients were considerably reduced. 126 Vactosertib, a TGF-β receptor kinase inhibitor, combined with nal-IRI plus 5-FU/leucovorin, suppressed tumor invasion via CCDC80 and improved overall survival in a murine pancreatic cancer model. 130 Taken together, these findings suggest that the use of TGF-β inhibitors may be beneficial in the treatment of cold tumors.
Breaking the complex physical barrier of the tumor to restore the activity of the antitumor immune system
The physical obstruction of aberrant tumor vasculature plays a major role in preventing T cells from penetrating the tumor to exert their antitumor immune action. The lack of vascular function, together with abnormal tumor vasculature formation, prevents the drug from reaching the tumor cells. Therefore, restoring the normalization of the tumor vasculature is a viable approach to restoring cold tumor immune activity. The use of antiangiogenic drugs can significantly contribute to the progression of tumor vascular normalization. 131 They can avoid vascular abnormalities, improve blood flow, reduce the hypoxic tumor environment and vascular permeability, and promote the recruitment of T cells in the TME. They can also induce upregulation of the leukocyte adhesion molecules ICAM-1 and VCAM-1 in tumors, favoring increased infiltration of T cells in the TME. 132 Currently, safety studies in patients with advanced solid tumors treated with bevacizumab (Avastin) to step up the dose of combretastatin and using a combination of Durvalumab, Bevacizumab, Tremelimumab, and transarterial chemotherapy (TACE) in patients with hepatocellular or biliary tract cancer have yielded favorable results.133,134
Inducing the release of chemokines from tumors to enhance the immunocidal effect of T cells
Following T-cell exudation, they need to be guided by chemokines to reach the tumor site and exert their antitumor effects. CXC chemokine receptor 3 (CXCR3) is principally distributed on CD4+ and CD8+ T cells. It comprises 3 ligands, CXCL9, CXCL10, and CXCL11. 135 The CXCL9, CXCL10, CXCL11/CXCR3 axis plays an essential role in regulating the migration, differentiation, and activation of immune cells and promotes the recruitment of NK cells, macrophages, and CTL in the immune system to exert powerful antitumor effects. Enhanced expression of CXCL9, CXCL10, and CXCL11 is, therefore, an ideal target for antitumor drug development, whereas high expression of CXCL10 has been found to be positively associated with better prognosis and overall survival in patients with rectal, osteosarcoma, and hepatocellular carcinoma.136,137 In addition, the CXCL12/CXCR4 axis also plays a decisive role in tumor progression, angiogenesis, metastasis, and survival. 138 Combining the CXCR4 antagonist BL-8040 (Motixafortide) with the PD-L1 inhibitor pembrolizumab and chemotherapy in patients with metastatic PDAC resulted in an increase in CD8+ T effector T cells. The combination also reduced the number of MDSCs and Tregs and promoted the restoration of the immune activity in the immunosuppressive TME, giving hope for the conversion of cold to hot tumors. 71
Nanoparticles combined with photothermal therapy to restore immune activity in the TME
Photothermal therapy (PTT), designed for tumors, is characterized by localized treatment and high controllability.73,139,140 It often utilizes nanomaterials (organic and full nanoparticles) as photothermal agents that are irradiated with near-infrared light, and the photosensitizer is excited by specific wavelengths to release energy to kill target cells and tissues.141,142 The PTT can also locally direct the activation of tumor-associated antigens to enhance the antitumor immune effect.143,144 Nanoparticles developed by combining PTT and immunogenic cell death inducers can exert powerful antitumor immune effects. 145 They can act as an antigen-loading platform carrying immunomodulators and tumor antigens that, when recruited by dendritic cells, exert an antitumor immune effect.146,147 Nanoparticle-based drugs can be divided into 3 distinct categories: targeting cancer cells to induce immunogenic cell death, 148 targeting the tumor immune microenvironment to activate immune cells (eg, CTL cells, macrophages) and promote their infiltration into the TME and exert antitumor immune effects, 7 and finally, targeting the peripheral immune system to restore the function of the peripheral immune system. Although nanoparticle-based antitumor therapies have achieved remarkable success, their use as a platform to restore the immune activity of the TME and exert an antitumor effect against immune-deficient cold tumors warrants further investigations. The short residence time of nanoparticles in human circulation prevents sufficient doses from penetrating tumor tissues to exert their effects, which is one of their limitations. 149
Restoration of T-cell mitochondrial activity to improve immunosuppression by TME
Mitochondrial hijacking of immune cells reduces the effectiveness of PD-1 antibodies in killing cancer cells. Conversely, mitochondrial activation of T cells can enhance the utility of PD-1 antibodies in the TME by improving recognition and supplying energy for sustained T cell activation. 61 Related studies have evinced that mitochondrial enhancers (AMPK activator, pGC-1α [peroxisome proliferator-activated receptor γ coactivator 1α], and mTOR [mechanistic target of rapamycin]) can increase the antitumor activity of anti-PD-1 antibodies. 150 Moreover, mitochondrial quality was shown to be partly improved by regular exercise, adequate sleep, healthy weight, a low SDA (specific dynamic action) diet, and smoking cessation. 151 Increasing mitochondrial levels within T cells may restore antitumor immune activity for a TME that is highly immunosuppressed by cold tumors and improve the efficacy of PD-1 antibodies.
Perspective
Restoring the immune activity of highly immunosuppressive TME in cold tumors remains one of the most pressing issues facing immunotherapy today. Cancer vaccines have achieved satisfactory results in prostate cancer, ovarian cancer, melanoma, non–small cell lung cancer, and bladder cancer; nonetheless, unequivocal efficacy has not been achieved in most cancer patients. There is an urgent need to explore suitable neoantigens as targets for advanced cancer patients with different cancer types and attempt personalized vaccine development in the future. Meanwhile, targeting the intestinal flora may be a promising approach for restoring the activity of the immunosuppressive TME in cold tumors. Further experimental data are necessitated to define the application of floral transplantation and eligible cancer patients, as well as identify which specific flora successfully elicits therapeutic responses. In addition, various measures were outlined to improve T-cell infiltration, such as anti-MDSC therapy, TGF-β inhibitors, antivascular therapy, chemokine antagonists, nanoparticles, and PTT. The use of immune checkpoint blocking therapy (ICBT) in combination with these therapies fractionally restored T-cell activity and improved the clinical outcomes of immunotherapy. However, more clinical data are still necessary to further evaluate the administration sequence and optimal dose of combination therapies and identify individuals for whom alternative combinations of therapies are indicated to maximize clinical benefits.
Conclusions
The advent of immunotherapy has provided additional therapeutic options and improved the survival rates of patients with advanced tumors. In response to the insensitivity of most cold tumors to ICBT therapies, a great deal of research is being conducted to restore the immune activity of cold tumors and convert them to hot tumors. These range from the development of drugs to intervene with immunosuppressive-related cytokines (eg, MDSCs and TGF-β) at the molecular level to the development of therapies to break down the physical barrier of the tumor at the tissue level and further attempts to restore the activity of the immune system by various means. Several studies regarding novel therapies such as oncolytic viruses, cancer vaccines, and nanoparticles in combination with PTT are underway. With a better understanding of the immunosuppressive mechanisms underlying cold tumors, we postulate that immunotherapy has a promising future for the treatment of advanced tumors.
Footnotes
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Training Project of Key Talents of Youth Medicine in Jiangsu province, China (No. QNRC2016330) and Graduate Research- Innovation Project in Jiangsu province (SJCX22_1816), the Key Disease Standardization Diagnosis and Treatment Project in Jiangsu province (N0.BE2015664), the Academic Science and Technology Innovation Fund for College Students (No. x20180714) the Social Development-Health Care Project of Yangzhou, Jiangsu Province (No. YZ2018087), the Social Development Project of Yangzhou, Jiangsu Province (No. YZ2021075), the Graduate Research and Practice Innovation Plan of Graduate Education Innovation Project in Jiangsu Province (N0. SJCX211644), and High-level talent “six one projects” top talent scientific research project of Jiangsu Province (No. LGY2019034), Social development project of key R & D plan of Jiangsu Provincial Department of science and technology (BE2022773). The funding bodies had no role in writing the manuscript.
Author Contributions
CW and YM drafted the manuscript. FW, YL, YC, and BZ researched the literature. QZ drafted figures. DW and DT critically revised the article for important intellectual content. All authors read and approved the final manuscript.