
Abstract
Introduction:
Haemoglobin H (Hb H) disease is an alpha thalassemia characterised by either 3 alpha-globin gene deletions (deletional type) or 2 alpha-globin gene deletions with 1-point mutation (nondeletional type). Haemoglobin H-Constant Spring thalassemia is the most common Hb H disease in Asia. Kikuchi-Fujimoto disease (KFD) is an important cause of prolonged fever in thalassemia and is often self-limiting.
Case Presentation:
A 30-year-old women of Malay ethnicity presented to the thalassemia unit with a month history of prolonged fever, headache, and painful enlarged neck lymph nodes. She is known to have Hb H-Constant Spring thalassemia, in which she is on 3-monthly blood transfusion. Physical examination revealed persistent pyrexia of 38°C. She had multiple tender bilateral cervical lymphadenopathies with the largest measuring 4 × 4 cm. The complete blood count revealed hypochromic microcytic anaemia with leucopenia and a normal platelet count. She had hyperferritinemia of 3500 ng/mL. The DNA analysis of alpha-globin gene showed heterozygosity for alpha zero thalassemia South East Asian deletion with termination codon mutation (TAA-CAA) which was consistent with Hb H-Constant Spring thalassemia. Numerous investigations for her prolonged fever including cultures did not yield any positive results. Whole-body computed tomography (CT) imaging showed diffuse lymphadenopathies and hepatosplenomegaly. Finally, a left cervical lymph node biopsy was performed which was consistent with KFD. She was treated with oral prednisolone which was gradually tapered based on response. Currently, she is asymptomatic and is in complete remission.
Conclusion:
Kikuchi-Fujimoto disease should be considered as a cause for prolonged pyrexia in a patient with thalassemia. An early diagnosis of KFD would avoid an unnecessary battery of investigations. This case highlights the importance of clinicopathological correlation in managing patients with thalassemia as these patients often have other associated morbidities.
Introduction
Haemoglobin H (Hb H) disease is an alpha thalassemia caused by either 3 alpha-globin gene deletions (deletional type) or the more severe type of 2 alpha-globin gene deletions with 1-point mutation (nondeletional type). Haemoglobin H-Constant Spring (Hb H CS) is the most common nondeletional type of alpha thalassemia in Asia. 1 Haemoglobin Constant Spring is named after the Constant Spring district in Jamaica. 2 Haemoglobin H CS, as compared to deletional Hb H disease, presents with more severe anaemia requiring regular blood transfusion, splenomegaly, and hyperferritinemia which is attributed to enhanced iron absorption by the gut and ineffective erythropoiesis. 3 Kikuchi-Fujimoto disease (KFD) should be considered in a patient presenting with unexplained prolonged fever with tender cervical lymphadenopathies. KFD is also known as histiocytic necrotizing lymphadenitis which was described by Kikuchi and Fujimoto in year 1972. 4 The course of this disease is often benign and self-limiting in nature although there have been reports of aggressive disease leading to fatality. There has been no evidence to suggest any causal or temporal relationship between thalassemia and Kikuchi-Fujimoto disease.
Case Presentation
A 24-year-old woman of Malay ethnicity presented to the thalassemia unit with a month history of prolonged fever, headache, and painful enlarged neck lymph nodes. She is known to have Hb H CS thalassemia, in which, she is on 3-monthly blood transfusion. She is on iron chelation subcutaneous deferoxamine 500 mg thrice weekly. She has a family history of Hb H disease. There is no history of parental consanguinity. She is a nonsmoker, a teetotaler, and works as a bank clerk.
Physical examination revealed a pale and jaundiced female with persistent pyrexia of 38°C. She was not septic looking and did not reveal any skin rash. She had bilateral palpable tender cervical lymphadenopathies with the largest measuring 4 × 4 cm. They were rubbery on palpation with no obvious nodal discharge or overlying skin colour changes. She had hepatosplenomegaly. Other systems were unremarkable.
The complete blood count revealed hypochromic microcytic anaemia, leucopenia, and a normal platelet count. She had hyperferritinemia of 3500 ng/mL. The other laboratory parameters are as tabulated in Table 1. Peripheral blood film (Figure 1) portrayed features consistent with hemoglobinopathy associated with chronic hemolysis. Capillary electrophoresis (Figure 2) showed Hb A of 69.1%, Hb A2 of 0.4%, Hb F of 0%, Hb H of 26.6%, Hb Barts of 1.5% and Hb CS of 2.4%. The agarose alkaline and acid gel electrophoresis (Figure 3A and B) revealed a faster-move band (Hb H band). The DNA analysis of alpha-globin gene showed heterozygosity for alpha zero thalassemia South East Asian (SEA) deletion with termination codon mutation (TAA-CAA) which was consistent with Hb H-Constant Spring thalassemia. The DNA analysis for Beta globin gene did not reveal any mutations.
Tabulation of laboratory parameters.


Peripheral blood film shows hypochromic microcytic erythrocytes, polychromasia, spherocytes, numerous target cells, and irregular contracted erythrocytes.

Capillary electrophoresis shows predominantly Hb A of 69.1%, Hb A2 of 0.4%, Hb F of 0%, Hb H of 26.6%, Hb Barts of 1.5% and Hb Constant Spring of 2.4%.
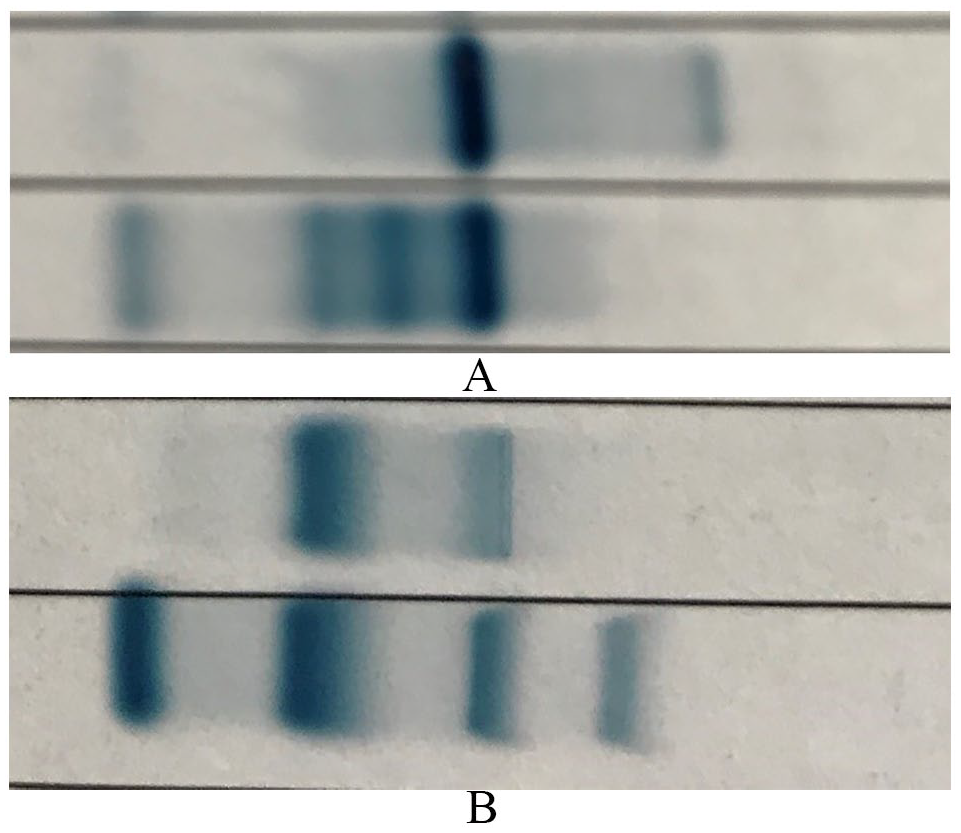
Agarose gel electrophoresis shows a faster-move band (Hb H band) on the (A) alkaline and (B) acid gel electrophoresis.
Blood, fungal, and tuberculosis cultures were sterile. A complete autoimmune panel was negative. An abdominal ultrasonogram and transthoracic echocardiogram did not reveal any microabscesses or vegetations. Magnetic resonance imaging (MRI) T2* of the cardiac and liver showed moderate liver iron overload and normal myocardial iron load. A whole body computed tomography (CT) imaging showed diffuse cervical, axillary, and abdominal lymphadenopathies with the largest lymph node measuring 4 × 4 cm.
A decision was made to perform a cervical lymph node biopsy. It showed paracortical expansion by histiocytes, small lymphocytes, and tingible body macrophages. The histiocytes had pale and crescentic nuclei. Extensive apoptosis with large areas of necrosis were present. There were no neutrophils, granulomas, or multinucleated giant cells seen. Immunohistochemical studies showed histiocytes positive for CD68 and myeloperoxidase (MPO). Most small lymphocytes were CD8+ T-lymphocytes. Periodic acid Schiff (PAS), Grocott’s methenamine silver (GMS), and Ziehl-Nielsen stains were negative for fungal bodies and acid-fast bacilli.
On the basis of these findings, a diagnosis of KFD was made. She was treated with oral prednisolone 1 mg/kg daily for a month followed by a gradual taper of 10 mg 2 weekly based on response. She responded clinically with complete resolution of symptoms. A repeat end-of-treatment whole body CT imaging showed complete resolution of lymphadenopathies. Currently, she is on a 3-monthly follow up at the thalassemia clinic.
Discussion
This patient had fever, headache, and tender cervical lymphadenopathies which are classical features of KFD. This disease affects young female Asians below the age of 40 and may present with skin rash over the face and upper body. 5 Those who present with cutaneous features often have a more protracted course of illness. Kikuchi-Fujimoto disease is diagnosed primarily by tissue morphohistology. Immunohistochemical stains are frequently not required to aid the diagnosis of KFD. Histologically, in KFD, lymph nodes have a partially preserved architecture with paracortical expansion and large areas of necrosis. Abundant crescentic histiocytes, clusters of plasmacytoid dendritic cells, and immunoblasts are visible at the edge of the necrosis. 6 Small lymphocytes, plasma cells, and tingible body macrophages are frequently present with neutrophils and eosinophils usually being absent on histology. 6
Many infective agents contribute to necrotizing lymphadenitis. Tuberculosis, histoplasmosis, infectious mononucleosis, Epstein-Barr infection, leprosy, human herpes virus, and syphilis are probable differential diagnoses. 7 Special stains are often useful to identify these infective agents. In our patient, PAS, GMS, and Ziehl-Nielsen stains of the tissue were negative for fungal bodies and acid-fast bacilli, thereby reducing the probability of a diagnosis of infection. Autoimmune disorders such as systemic lupus erythematosus (SLE) share very similar features to KFD. As the patient in this case is in the young reproductive age group, SLE should always be considered as an important differential diagnosis. Kikuchi-Fujimoto disease may be a clinical feature or an incomplete phase of lupus lymphadenitis. 8 However, KFD could occur without the co-presence of SLE, thus suggesting that SLE and KFD are 2 independent entities which may coexist in vulnerable subjects. It is often challenging to differentiate SLE from KFD based on histology. The presence of hematoxylin bodies (clusters of basophilic materials within the lymph node sinuses) and total absence of plasma cells are characteristic for SLE. 8 Malignant lymphomas such as Hodgkin lymphoma, T and B non-Hodgkin lymphoma also mimic KFD. Immunohistochemistry plays an imperative role in such cases. Hodgkin lymphoma is usually characterised histologically by the presence of large binucleated Hodgkin and Reed Sternberg (HRS) cells with a cellular background comprising of lymphocytes, neutrophils, eosinophils, plasma cells, and histiocytes. Furthermore, tumour cells in Hodgkin lymphoma frequently stain positive for CD15, CD30, and PAX5 on immunohistochemistry. Non-Hodgkin lymphomas depict complete architectural effacement, a higher proliferation mitotic index and the absence of reactive histiocytes on histology. 8 Immunohistochemistry plays an important role in delineating the subtypes of non-Hodgkin lymphoma.
Chronic inflammatory disorders such as Kimura disease is often a differential diagnosis to consider in a patient who presents with unexplained lymphadenopathies. Kimura disease is characterised by the presence of germinal centre hyperplasia with intragerminal fibrosis and proteinaceous material. 9 Frequently, there is an increase in paracortical plasma cells, folliculolysis, and interfollicular eosinophils. 9
Kikuchi-Fujimoto disease is usually self-limiting and may resolve spontaneously in 1 to 4 months without any treatment. Severe cases may benefit from oral or intravenous pulses of corticosteroids, intravenous immunoglobulin, and hydroxychloroquine. 10 Patients with coexistent KFD and SLE may have a more protracted course and should be treated aggressively with steroids and hydroxychloroquine.
Conclusion
Kikuchi-Fujimoto disease should be considered as an imperative cause of prolonged unexplained pyrexia in a patient with thalassemia. An early diagnosis of KFD would avoid an unnecessary battery of investigations. This case highlights the importance of clinicopathological correlation in managing patients with thalassemia as these patients often have other associated morbidities.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was self-funded by the authors.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
G.K. analysed the data, designed the paper, wrote the manuscript, made critical revisions and approved the final version.
Informed Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.