
Abstract
Background:
Accumulating evidence has demonstrated that epithelial-mesenchymal transition (EMT) plays a critical role in breast cancer (BRCA) initiation, invasion, metastasis, and prognosis.
Objectives:
To develop and validate a comprehensive EMT-related gene signature for robust prognosis prediction in BRCA.
Design:
Retrospective multi-cohort study.
Methods:
We obtained 1223 BRCA samples from The Cancer Genome Atlas (TCGA) and 1184 EMT-related genes from the dbEMT2.0 public database. Prognostic genes were selected via univariate Cox and LASSO regression analyses to construct a risk score model, which was subsequently validated in independent internal cohort (TCGA) and external cohorts (UCSC and GEO). Finally, a nomogram integrating the risk score with clinical parameters was established.
Results:
A 15-gene EMT signature was identified and used to stratify patients into high- and low-risk groups. The high-risk group exhibited significantly poorer overall survival in the TCGA cohort (P < .05), a finding consistently validated across 4 independent datasets (all P < .05). The risk score served as an independent prognostic factor (hazard ratio = 2.386, P < .001). The integrative nomogram, incorporating the risk score, age, and N and M stages, demonstrated moderate accuracy for survival prediction (C-index = 0.711).
Conclusions:
We developed and validated a novel 15-gene EMT signature and a corresponding nomogram, which provide a potential tool for prognostic stratification in BRCA patients.
Introduction
Breast cancer (BRCA) is the most common cancer and second leading cause of cancer-related mortality in women worldwide. 1 Due to the convenience of imaging studies and surgical biopsy, BRCA has a higher detection and early intervention ratio. An integrated treatment system for BRCA, including surgery, chemotherapy, radiotherapy, endocrine therapy, and targeted therapy, has resulted in a better survival prognosis.
However, the treatment efficacy of BRCA remains suboptimal when metastasis occurs. 2 In this process, cells with mesenchymal attributes show a predominance, undergo epithelial-mesenchymal transition (EMT), and present the characteristics of poorly differentiated cancer cells. 3 EMT is a dynamic process that is essential for embryonic development, wound healing, and tumor progression. 4 Under special physiological or pathological conditions, EMT alters the adhesion ability, polarity, and differentiation characteristics of epithelial cells, facilitating their migratory and invasive. 5 This process is always accompanied by decreased expression of E-cadherin; overexpression of N-cadherin, Vimentin, and matrix metalloproteinases; and relocation of β-catenin from the cytomembrane to the nucleolus. 5 Several transcription factors such as Snail, Twist, and Zeb have been shown to induce EMT.5,6 Specific extracellular signals activate EMT, which then provokes epithelial cell reprogramming toward a mesenchymal phenotype, 6 leading to acquired migration and self-renewal ability, fostering the formation of secondary tumors at distant sites. 7 Several independent EMT-related factors have been reported to be critical for distant metastasis in BRCA; however, the underlying mechanisms remain unclear.
The functions of EMT cover various domains in tumors, including tumor metastasis initiation as well as malignant progression, tumor stemness, intravasation to the blood, and resistance to therapy. 8 In recent years, studies have suggested that EMT is not a unilateral binary process with 2 distinct cell populations, epithelial and mesenchymal, 9 but is triggered in a gradual manner characterized by multiple cellular states expressing different levels of epithelial and mesenchymal markers and exhibiting different functional characteristics between epithelial and mesenchymal cells. 10 The complexity and plasticity increase the difficulty of segmentation research and single-biomarker screening in metastatic BRCA. 11 Therefore, it is necessary to explore the original gene signature by taking EMT as a whole.
Existing multi-gene prognostic assays, such as Oncotype DX and PAM50, 12 primarily prioritize pathways related to proliferation, hormone receptors, 12 and HER2 signaling. 13 In contrast, EMT represents a fundamental and early process underlying cancer metastasis and therapeutic resistance. Consequently, EMT-related gene expression may capture complementary prognostic information, specifically regarding invasive and metastatic potential. 7 Although prior studies have frequently focused on individual EMT transcription factors, prognostic models derived from a systematic integration of the EMT gene landscape remains underexplored. Therefore, we hypothesized that a prognostic model based on a comprehensive set of EMT-related genes could provide a robust and independent tool for predicting overall survival (OS) in BRCA patients.
In the present study, we constructed a 15-gene risk score model by analyzing 3 prime public databases: The Cancer Genome Atlas (TCGA), UCSC Public Hub, and Gene Expression Omnibus (GEO). While the reliability and independence of this novel model were confirmed, we also established a nomogram to assess the overall survival (OS) of each BRCA patient, aiming to assist in risk stratification.
Materials and Methods
Data Collection and Preprocessing
A flowchart of the present study is shown in Figure 1. A total of 1223 samples were included in this study, and data including RNA-seq matrix and clinical parameters were downloaded from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/). Selected cohorts of breast cancer (Chin 2006) were downloaded from the UCSC Public Hub (https://xenabrowser.net/hub/). GSE21653, GSE42568, and GSE48408 were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). Generally, raw data should meet the following criteria: (1) genes were excluded if normalized expressions were zero in over half of the samples; (2) genes with missing expression values in more than 50% of the samples were deleted; (3) genes whose expression was invariable or minimally variable were removed; (4) samples without necessary clinical parameters or OS = 0 were eliminated; and (5) normal adjacent samples could be ruled out after differential analysis. EMT-related genes were obtained from a public database (dbEMT2.0: http://dbemt.bioinfo-minzhao.org/). 14 Given that the databases were open access, ethical approval from an ethics committee was not required.
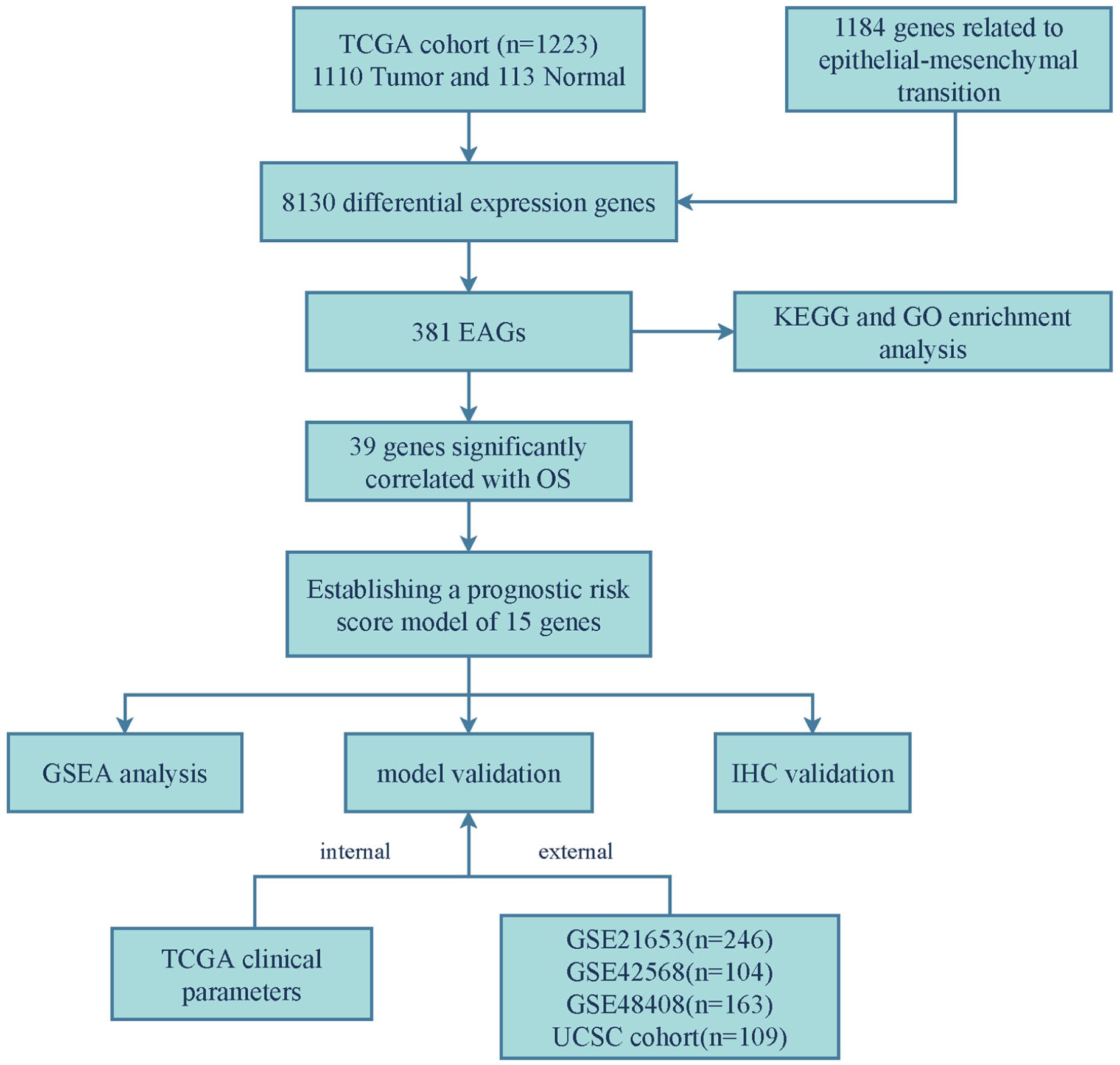
Flowchart of the present study.
To eliminate potential technical batch effects, differential expression analysis within TCGA, the ComBat algorithm (sva R package) was applied. For external validation across independent cohorts (UCSC and GEO datasets), the pre-defined risk-score formula was applied to each cohort separately, followed by cohort-specific survival analysis. This validation strategy inherently avoids cross-platform batch effects.
Identification and Functional Analysis of EMT-Associated Genes (EAGs)
Differential gene expression analysis was performed based on the TCGA database using the R package “edge R.” Specifically, we used the Benjamini-Hochberg method to adjust the P-value (adj P-value < .05, |log FC|> 1), and then obtained EAGs from the dbEMT2.0 database by intersection of the above results in the Venn diagram. KEGG and GO functional enrichment analyses were performed using the “clusterprofiler” package in R software. The protein-protein interaction (PPI) network was analyzed using the STRING online database (https://string-db.org/), and sub-networks and hub modes of the PPI network were extracted using the Cytoscape software.
Construction of an EAGs Risk Score Model for Prognosis Prediction
The expression matrix of these EAGs was used to establish a risk-score model. First, a univariate Cox proportional risk model was applied to estimate the impact of each EAG on OS, and a P value <.01 was considered statistically significant. Second, LASSO-Cox regression was employed to narrow down the genes and strengthen the stability of the model. This analysis was implemented using the “glmnet” R package with 10-fold cross-validation. The optimal penalty parameter (lambda, λ) was selected based on the minimum value of the partial likelihood deviance (1-standard-error criteria). Finally, the risk score was calculated using the formula:
Independent Prognostic and Survival Analysis of EAGs Risk Score Model
The risk score for each sample was calculated by using a specific formula. The proportional hazards assumption was verified using Schoenfeld residuals (via the cox.zph function in R), with no significant violations observed (all P > .05). Integrating clinical parameters, multivariate Cox regression analysis was performed to evaluate the independence of the risk score. Uniting the clinical parameters, multivariate Cox regression analysis substantiated the independence of “risk score” for prognosis prediction. The samples were then divided into high- and low-risk groups based on the optimal cutoff of the risk score, which was estimated using the “surv_cutpoint” (from the “survminer” R package) that maximized the survival difference assessed by the log-rank test. The Kaplan-Meier (K-M) survival curve was applied to show the OS of each group by using the log-rank test to compare the survival difference between the high-risk and low-risk groups. Time-independent receiver operating characteristic (ROC) curve analysis was used to calculate the area under the curve of 1-year, 3-year, and 5-year OS.
Validation of EAGs Risk Score Model by UCSC Cohort and GEO Cohorts
To prove the prognostic value of our risk score model, 1 UCSC cohort and 3 GEO cohorts (GSE21653, GSE42568, and GSE48408) were selected. The risk score of each sample was analyzed using the above-mentioned formula, and then these 4 validation cohorts were divided into high- and low-risk groups using the same method used in TCGA database validation. Critically, to ensure a rigorous external validation, the identical risk score formula and the gene coefficients derived from the TCGA training cohort were applied directly to all validation cohorts without any recalibration or retraining. The distribution and correlation between the risk scores, heatmaps, and survival data were visualized.
Establishment of a Prognosis Predictive Nomogram
Univariable and multivariable Cox regression analyses were used to calculate the influence of variables in TCGA cohort, including gene expression and clinical data. Previously, the risk score model was confirmed to be an independent factor; however, clinicopathological parameters such as age and pathological T, N, and M stages were also added. Variables that were statistically significant in univariate Cox regression were used in multivariable Cox regression analysis. The proportional hazards assumption underlying the Cox model used to construct the nomogram was assessed with Schoenfeld residuals and was satisfied (all P > .05). Based on multivariable Cox regression analysis, a nomogram integrating clinicopathological parameters with risk score was presented. Then the founded nomogram can be used to analyze the total points of each patient for predicting OS.
Gene Set Enrichment Analysis
Gene set enrichment analysis was conducted for searching the significantly enriched pathways between high-risk group and low-risk groups. Given that EMT is a comprehensive, multi-level biological process involving tumor migration, invasion, and complex molecular reactions, we utilized the 106 founder gene sets that constitute the “HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION” collection in MSigDB (https://www.gsea-msigdb.org/gsea/msigdb). This approach allows for a detailed evaluation of the specific functional cascades and interconnected pathways underlying the EMT spectrum within our risk model, providing a broader analytical context. 15 The significance of enrichment was assessed through 1000 permutations of the gene set. A gene set was considered significantly enriched if it met the following thresholds: |Normalized Enrichment Score (NES)| > 1, Nominal P-value (P value) < .05, and False Discovery Rate q-value (FDR) < 0.25.
Preliminary Assessment of Protein Expression Patterns
Immunohistochemical (IHC) staining images of these genes from the risk score model were downloaded from the Human Protein Atlas (HPA) database (https://www.proteinatlas.org/) for exploratory and descriptive purposes. Staining intensity was compared between representative images of normal breast tissue and BRCA tissue available in the HPA. The HPA-provided annotations (eg, “Not detected”, “Low”, “Medium”, “High”) were referenced to aid this qualitative assessment. To quantitatively assess protein expression, IHC staining analysis for candidate proteins was performed by Image J software. The IHC Scores (range 0-300, calculated as Σ (intensity × % of cells)) were evaluated for Semi‑quantitative analysis. Statistical comparison between normal and tumor tissues was performed using the Wilcoxon rank-sum test. This approach does not constitute a true experimental validation but serves as an initial observation to inform future investigations.
Reporting Guideline
The reporting of this study conforms to the TRIPOD + AI statement 16 (Supplemental Table 3).
Results
Patients’ Characteristics
A total of 1110 BRCA samples and 113 adjacent normal samples were downloaded from the TCGA-BRCA dataset and 1184 EMT-related genes were obtained from the dbEMT2.0 database (Supplemental Table 1). The clinicopathological characteristics included age, T stage, N stage, M stage, survival time, and survival status. The cohorts of Breast Cancer from UCSC (Chin 2006) (n = 109), GSE21653 (n = 246), GSE42568 (n = 104), and GSE48408 (n = 163) datasets were screened for external validation. Genes and samples that did not meet these criteria were also excluded.
To assess baseline comparability, we performed Wilcoxon rank-sum tests. No statistically significant differences were observed in Age, T stage, N stage, or M stage distribution between the model and validation cohorts (all P > .05), indicating a balanced distribution of baseline characteristics (Table 2).
EAGs Identification
First, gene probes in the mRNA-seq matrix were transformed to the corresponding gene symbols using the public database Ensembl (http://uswest.ensembl.org/index.html). A total of 39742 genes from 1110 BRCA samples and 113 adjacent normal samples were collected from the TCGA-BRCA dataset, and differentially expressed genes were obtained based on “edge” R package (adj P value < .01 and |log FC|> 1). There were 8130 differentially expressed genes between BRCA samples and normal samples, the expression patterns across individual samples were shown in a heatmap, with 2520 down-regulated and 5610 up-regulated (Figure 2A). The magnitude and statistical significance of these expression changes were summarized in the volcano plot, which highlighted the most differentially expressed genes (Figure 2B). In addition, 1184 EMT-related genes were obtained from the dbEMT2.0 database. A Venn diagram was used to identify 381 overlapping genes between the 2 datasets (Figure 2C), with 213 up-regulated and 168 down-regulated in BRCA samples compared with adjacent normal tissues. Next, KEGG and GO analyses were applied to explore the potential molecular mechanisms and biological functions of the 381 identified EAGs. The top pathways and terms directly correspond to the hallmarks of EMT. Specifically, KEGG pathways such as “ECM-receptor interaction” and “Focal adhesion” underpinned the altered cell adhesion and increased migration during EMT (Figure 3A). Concurrently, GO terms like “regulation of epithelial cell proliferation” reflected the accompanying loss of epithelial stability (Figure 3B). This focused profile confirms that our EAG signature captures the core functional network of EMT in breast cancer. Third, the STRING online website was used to visualize the interaction between the 381 EAGs (Figure 3C), with the following settings: (1) minimum required interaction score = 0.900 and (2) disabled structure previews inside bubbles and hide disconnected nodes in the network. With the use of “MCODE” in Cytoscape software, 6 sub-networks were separated (Figure 3D), which can be regarded as hub modes in the intricated networks.

EAGs identification. (A) Heatmap displayed the expression of 8130 screened differentially expressed genes between BRCA tumor samples (red annotation bar) and adjacent normal samples (blue annotation bar). (B) Volcano plot displayed 8130 significant DEGs (5610 upregulated in red; 2520 downregulated in blue) between tumor and normal tissues, based on thresholds of |log FC| > 1 and adj. P < .05. (C) Venn diagram with the interaction between TCGA-diff genes and EMT-related genes.
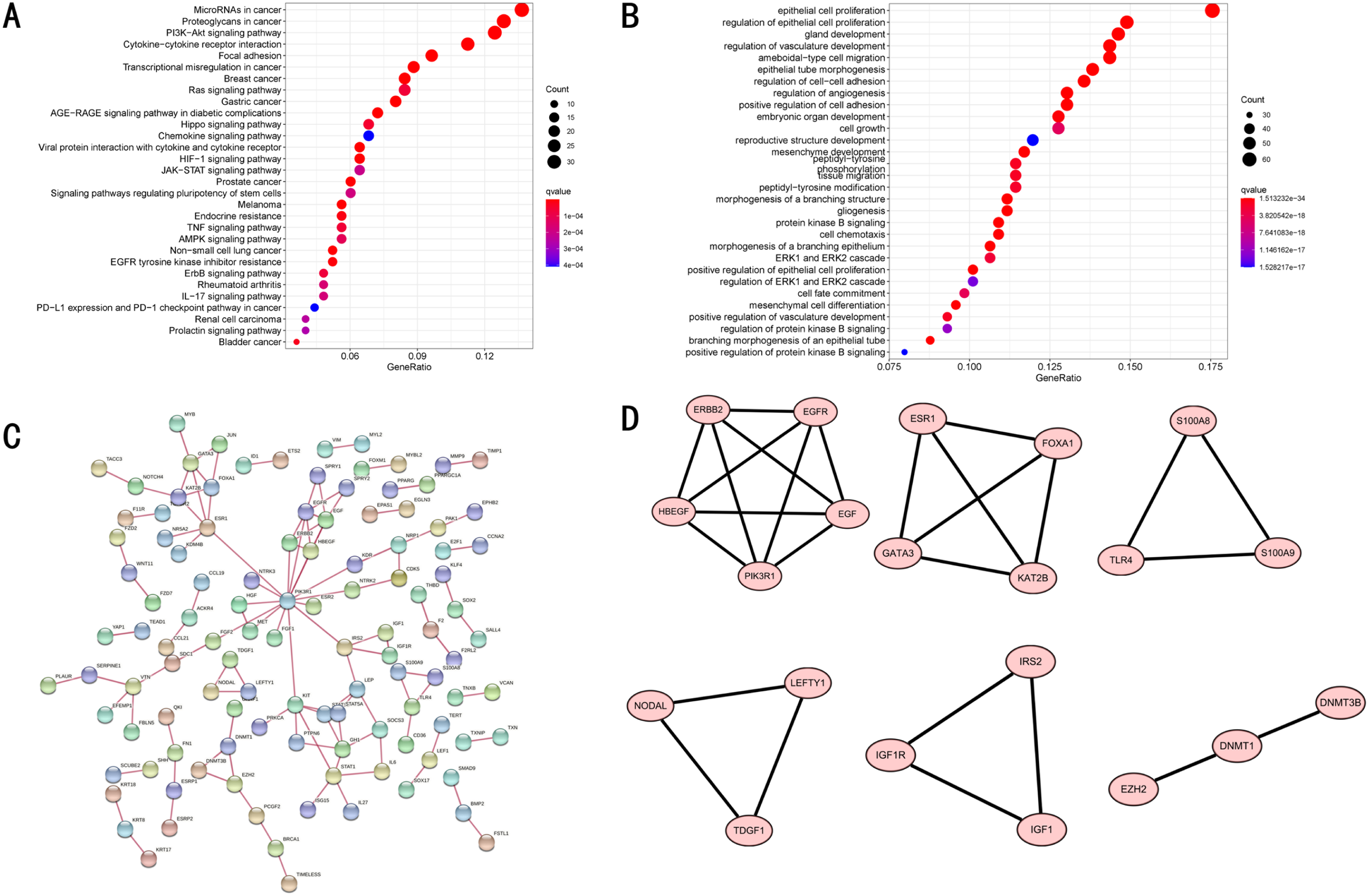
Functional enrichment analysis of the 381 EAGs. (A) KEGG enrichment analysis showing the first 30 pathways. (B) GO enrichment analysis showing the first 30 pathways. (C) PPI network showing the protein interaction between the 381 EAGs. (D) Six hub gene modes of the PPI network.
Constructing an EAGs Prognostic Risk Score Model
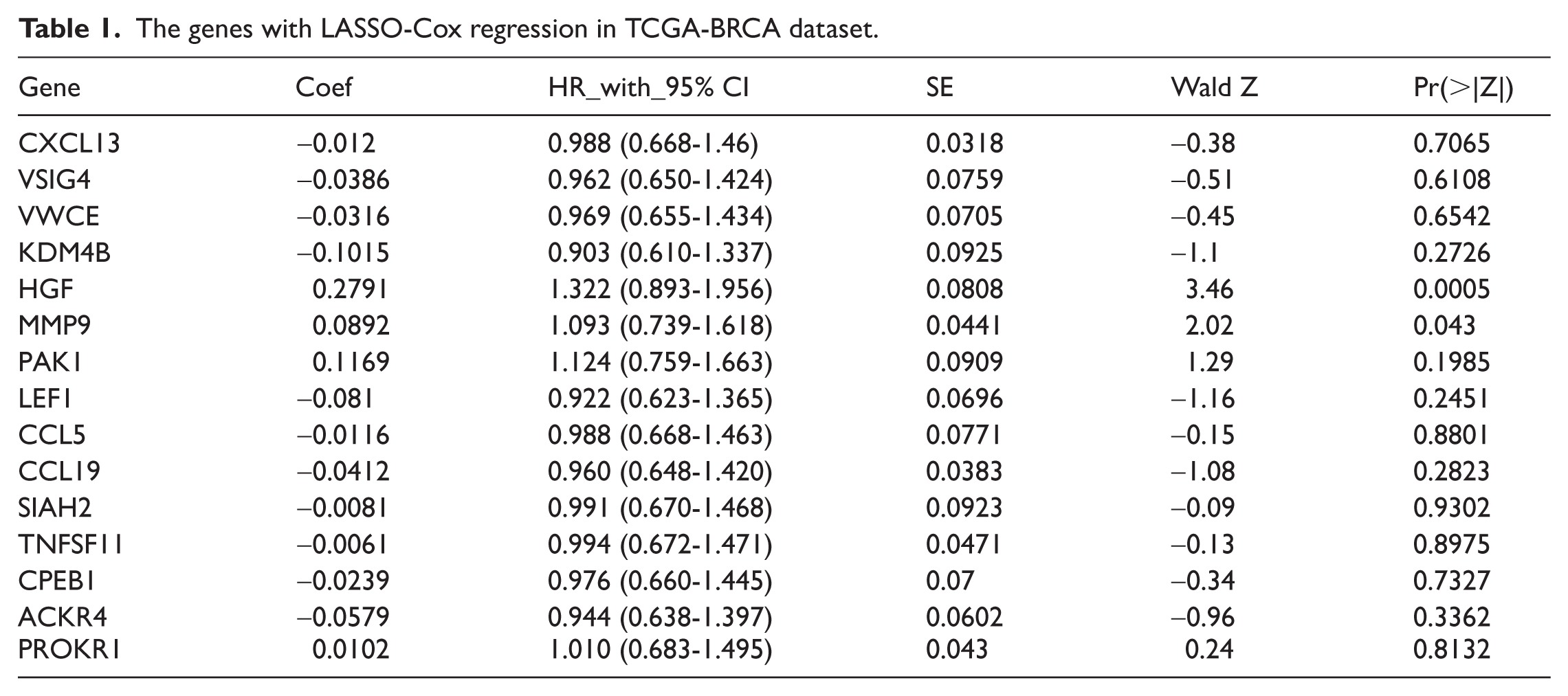
Univariate Cox regression analysis was performed for the 381 EAGs. A total of 39 genes (Supplemental Table 2) visualized by a forest map were identified as risk factors for the OS of BRCA (Figure 4A), and 15 genes (Table 1) were finally obtained using the LASSO-Cox regression analysis algorithm to narrow the number of significant genes and gain their coefficients (Figure 4B and C). The risk score for each patient in the TCGA cohort was calculated according to the formula described in the Methods section. Based on the median risk score, patients were stratified into high-risk and low-risk groups for subsequent analyses. We then checked the genetic alterations of these 15 genes from the novel risk score model for deeper research using the cBioPortal for Cancer Genomics website (http://www.cbioportal.org/) (Figure 5A). We obtained a differential expression diagram for other cancers (Figure 5B). In particular, the gene signature had a relatively high hazard ratio (HR) in gastric cancer and lung adenocarcinoma but a relatively low HR ratio in BRCA, head and neck squamous cell carcinoma, and cholangiocarcinoma, indicating that EAGs may have a significant value in other cancers.

Identification of risk factors for OS. (A) A total 39 genes related to OS of BRCA samples were shown in the forest diagram, as well as the Hazard ratio of each gene. (B) LASSO-Cox regression coefficient profiles of survival-related genes. (C) Cross-validation in the LASSO-Cox regression model to choose the tuning parameter. The first black dotted line indicated the optimal lambda.
The genes with LASSO-Cox regression in TCGA-BRCA dataset.
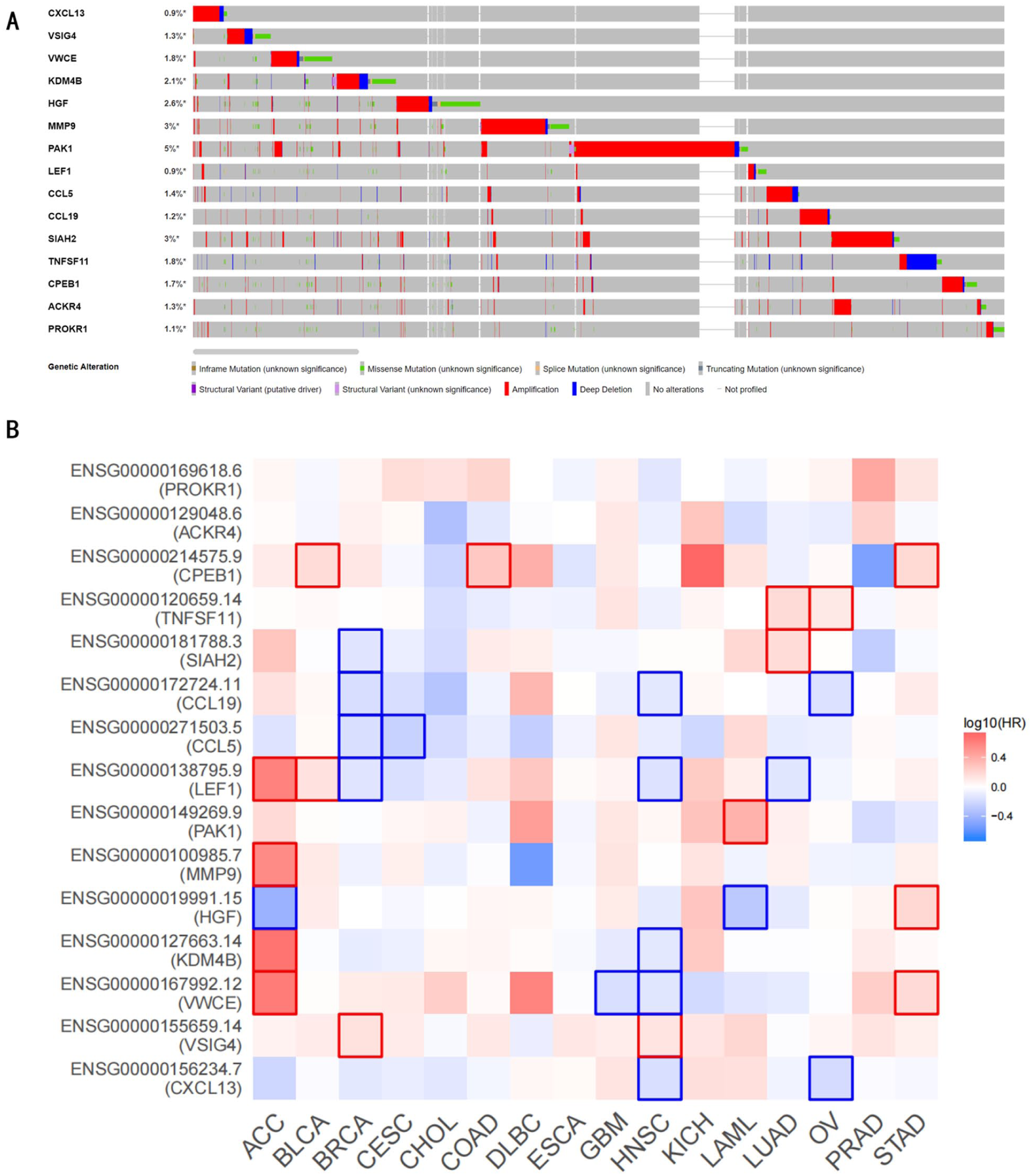
Deeper research of these 15 genes. (A) Genetic alteration of the 15 prognostic genes in TCGA-BRCA dataset. (B) Differential expression of these 15 prognostic genes in other types of cancer.
Integral and External Validation
According to the model, the BRCA samples were divided into high- and low-risk groups using the value as the cutoff point, which indicates the best sensitivity and specificity. The risk scores were positively correlated with the survival status of the BRCA samples in a linear graph, and the scatter plot showed that the low-risk group had more alive samples and longer survival time (Figure 6A and B), The differential expression of these 15 genes between the high-risk and low-risk groups is represented in a heatmap (Figure 6C). Moreover, K-M survival curve suggested that patients with high-risk scores showed lower OS, with respect to patients with low-risk scores (Figure 6D). The ROC curve at 1 year, 3 years, and 5 years was 0.65, 0.59, and 0.68, respectively (Figure 6E), indicating a modest discriminative ability.
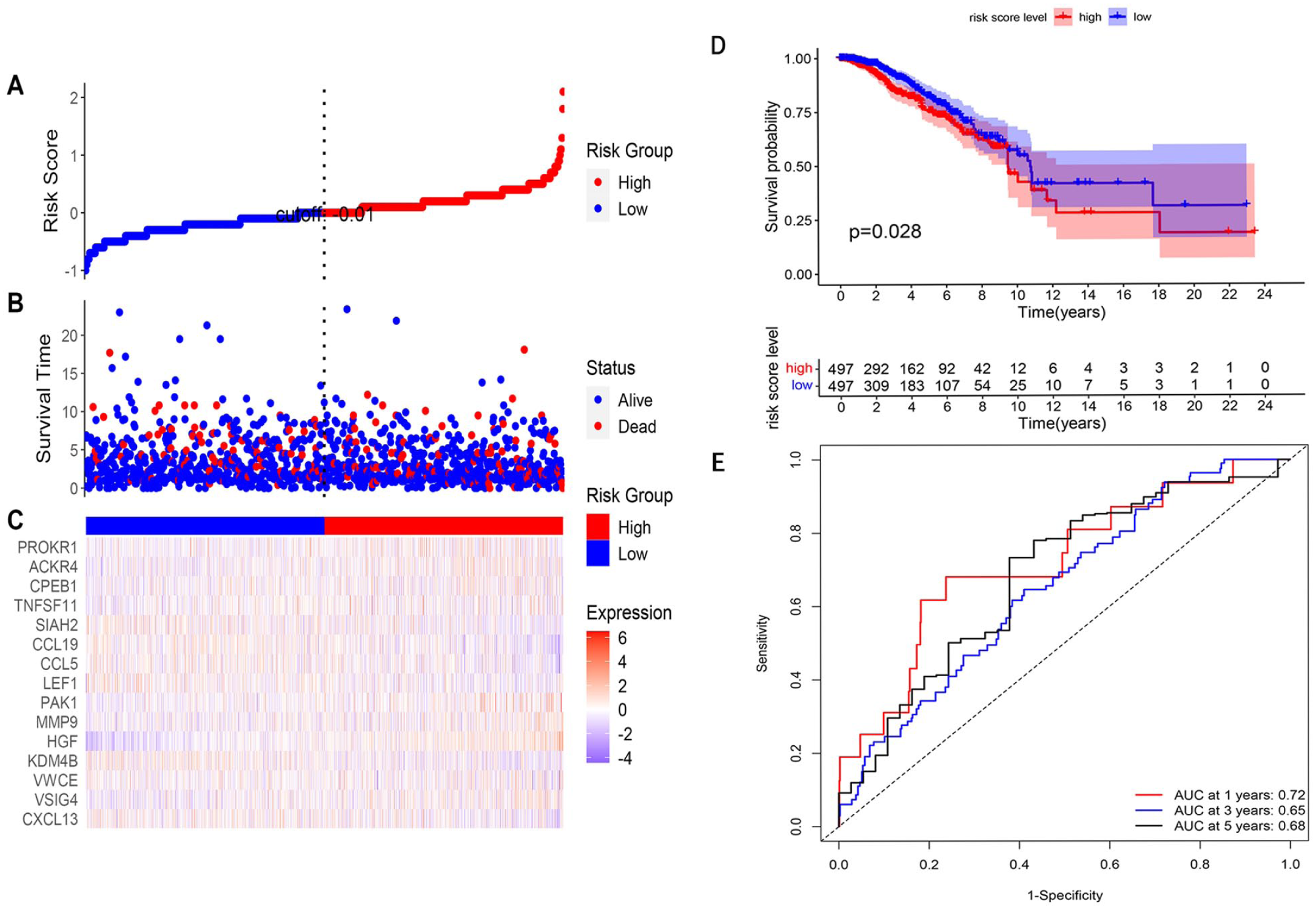
Integral validation. (A) According to the median risk score, cutoff point was identified as −0.01 in the liner graph. (B) The distribution of risk scores of BRCA samples, these dots present each sample’s survival status, blue dots present alive, and red dots present dead. (C) A heatmap of 15 LASSO-Cox genes between high-risk and low-risk group. (D) K-M analysis between high-risk and low-risk group in TCGA cohort (P < .05). (E) Time-dependent ROC curves of the prognostic gene signature in TCGA cohort.
The stability of this risk score model was further evaluated using 1 UCSC cohort and 3 GEO datasets (GSE21653, GSE42568, and GSE48408). Detailed information about the model and validation cohorts is shown in Table 2. The results were similar to integral validation in the TCGA database, with P values of <.001, <.001, .003, and <.001 in the UCSC, GSE21653, GSE42568, and GSE48408 cohorts, respectively. The ROC curves displayed the efficiency of 1-year, 3-year, and 5-year OS in each dataset (Figure 7).
Information of model cohort and validation cohort.

External validation. The distribution of risk scores (A), the heatmap of 15 genes in the model (B), the K-M curve of OS (C), and the ROC curve of accuracy (D) in UCSC cohort, GSE21653, GSE42568, and GSE48408 dataset.
To evaluate the performance of our EMT-based risk score within different biological contexts, we analyzed its distribution and prognostic value in the major molecular subtypes (Luminal A, Luminal B, HER2-enriched, TNBC) in the TCGA cohort. Interestingly, although the risk score distribution did not exhibit statistically significant differences among the 4 subtypes (Wilcoxon rank-sum test, P > .05; Supplemental Figure 1A), its prognostic utility was highly subtype-dependent. Stratified KM curves revealed that the risk score distinguished OS within the TNBC subgroup with borderline significance (log-rank test, P < .05), whereas no significant prognostic differentiation was observed in Luminal A/B or HER2-enriched subtypes (all P > .05, Supplemental Figure 1B). This suggested that the prognostic value of our EMT signature might be relevant in the more aggressive, mesenchymal-like TNBC subtype. However, as this was based on a single cohort, independent validation in larger TNBC datasets is necessary.
Establishment of a Prognosis Predictive Nomogram
Univariate and multivariate Cox regression analyses were performed to validate the prognostic value of the risk-score model. Univariate Cox regression analysis showed that the risk score and clinicopathological parameters (age, T stage, N stage, and M stage) were significantly related to the OS of patients with BRCA (Figure 8A). Then these variables were devoted in multivariate Cox regression analysis, ensuring that “risk score” was an independent prognostic factor (Figure 8B). To build a predictive method, a prognostic nomogram was generated to predict the 1-year, 3-year, and 5-year OS of patients with BRCA (Figure 9A). For instance, when the total number of points was 100, the 1-, 3-, and 5-year OS rates were 0.979, 0.856, and 0.579, respectively, where a higher total points score indicated a poorer prognosis and a greater risk of death. The C-index of this model was 0.711, indicating a medium accuracy. Calibration curves showed the prediction value of the nomogram and demonstrated high accuracy of the predicted survival at 1 year, 3 years, and 5 years (Figure 9B to D).
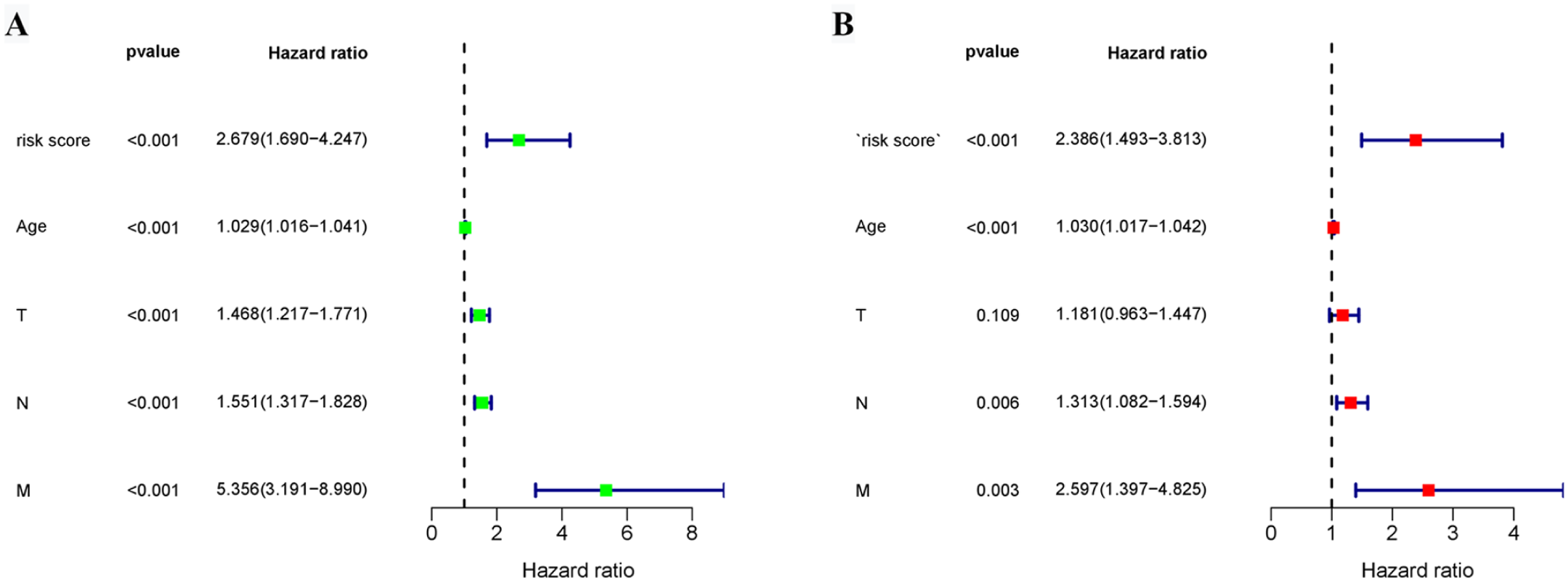
Cox regression analysis. Forest diagrams showing the univariate Cox regression analysis (A) and multivariate Cox regression analysis (B) in TCGA-BRCA cohorts.

Constructing nomogram. (A) Nomogram diagram showing the OS of 1 year, 3 years, 5 years, including risk score and other significant clinical parameters. The calibration curves of 1 year (B), 3 years (C), and 5 years (D) between nomogram and ideal model.
To directly evaluate the link between the EMT-based risk score and metastasis, we analyzed its association with pathological M stage in the TCGA cohort. Patients with distant metastasis (M1, n = 20) had significantly higher risk scores than those without metastasis (M0, n = 974) (Wilcoxon rank-sum test, P < .05). Furthermore, among M0 patients, a high-risk score remained predictive of worse OS (log-rank test, P < .05), suggesting a potential association with an elevated risk of future metastasis. (Supplemental Figure 2).
Gene Set Enrichment Analysis
After being divided into high-risk and low-risk groups, we investigated the enriched pathways using the 106 founder gene sets of the EMT hallmark. In the high-risk group, we observed a trend toward enrichment in the extracellular region (NES = −1.26, Nominal P-value < .001), however, this did not reach our pre-defined statistical significance threshold (FDR = 0.3829). In contrast, the low-risk group showed significant enrichment in several specific pathways derived from these founder sets, such as the GNF2 signature (all NES > 1, Nominal P-value < .001, FDR < 0.001) and VEGFA signature (all NES > 1, Nominal P-value < .001, FDR < 0.01). (Supplemental Figure 3). While preliminary, these enrichment patterns may hint at underlying biological differences, with the low-risk group potentially exhibiting features closer to epithelial stability, though further functional validation is required to substantiate these exploratory findings.
Protein Expression Patterns Based on HPA Database of the EMT Risk Score Genes
To determine the protein expression characteristics of these EMT risk score genes, immunohistochemical images from the HPA database were analyzed. We prioritized genes from the risk model based on 3 practical criteria: (1) a relatively higher absolute coefficient weight in the model, (2) the availability of clear and representative immunohistochemical staining images in the HPA database, and (3) the presence of comparable antibody staining in both normal breast and BRCA tissues to allow for discernible contrast. Based on these criteria, 8 genes (CXCL13, CCL5, SIAH2, MMP9, VWCE, PAK1, HGF, and PROKR1) were selected for comparative analysis. We compared the staining intensity between normal breast tissues and BRCA tissues to assess the differences. Semi-quantitative analysis using ImageJ‑derived IHC scores confirmed that higher protein levels for these 8 genes in BRCA tissues compared with normal breast tissues (Wilcoxon rank-sum test, all P < .05) (Supplemental Figure 4). This observation derived from a qualitative and semi‑quantitative review of public data, aligned directionally with our transcriptomic findings and might warrant future targeted validation.
Discussion
Despite better overall prognosis, advanced BRCA remains challenging due to recurrence, metastasis, and therapeutic resistance. 17 Since EMT is the initial step to distant metastasis in malignancy, 4 in which the epithelial phenotype can be transformed into a mesenchymal phenotype, accompanied by the dynamic alteration of biomarkers and EMT transcription factors, then activating the processes of biological signal transduction. 18 The plasticity of epithelial cells and reprogramming of signatures in biological metabolism provide theoretical evidence for this study. This study focused on developing a molecular prognostic model based on EMT gene expression, and providing a tool for refined risk assessment in breast cancer.
In this study, a prognostic risk score model, including 15 genes, was constructed using univariate Cox regression and LASSO-Cox regression based on 381 differentially expressed EAGs. The risk score model consisting of CXCL13, VSIG4, VWCE, KDM4B, HGF, MMP9, PAK1, LEF1, CCL5, CCL19, SIAH2, TNFSF11, CPEB1, ACKR4, and PROKR1 was verified to be efficient in predicting the prognosis of BRCA. It was noteworthy that the roles of some constituent genes, such as VWCE and PROKR1, in cancer biology were complex and context-dependent. While VWCE had been reported as a potential tumor suppressor in breast cancer, 19 PROKR1 is often associated with oncogenic functions in other malignancies. 20 These observations highlight the intricate and sometimes pleiotropic nature of genes within the EMT network, and further investigation is warranted to clarify their precise roles in breast cancer progression. A nomogram was established, which displayed high accuracy in the calibration curves, and the risk score with a high HR value (HR = 2.386) played an important role in OS prediction. Finally, TCGA cohort was divided into high-risk and low-risk groups for gene set enrichment analysis based on the 106 founder gene sets of the EMT hallmark. Interestingly, while not reaching strict statistical significance, the high-risk group showed a trend toward enrichment in the “extracellular region part” pathway, suggesting a potential role for transmembrane proteins and extracellular matrix remodeling in this aggressive phenotype, though further validation is required.
Our study introduced a distinct EMT-based prognostic model for BRCA, with several strengths. First, we applied LASSO-Cox regression to develop a concise 15-gene signature that was less prone to overfitting and more suitable for potential clinical use. Second, the model’s robustness was confirmed across multiple independent patient cohorts (TCGA, UCSC, and GEO datasets), which differed in patient backgrounds and testing platforms. Third, we combined the risk score with common clinical factors in a practical nomogram, making it easier to estimate individual patient survival. Forth, our gene signature reflects not only core EMT activity but also related processes such as extracellular matrix remodeling, immune response, and metabolism, offering a more integrated view of cancer progression than single-pathway models. Notably, while the risk score did not vary significantly in 4 molecular subtypes, its prognostic relevance was specifically observed in the TNBC subgroup. This specificity aligns with the characteristically aggressive and mesenchymal-like phenotype of TNBC, suggesting that the EMT biology captured by our model is particularly salient and clinically actionable in this high-risk population.
Recently, Mo et al 21 constructed an EAGs prognostic model in colorectal cancer and combined the risk score with clinical parameters, including age, sex, and TNM staging system. However, there were a few common genes between our gene signatures and Mo’s, suggesting that the EMT-contributing genes are tumor-specific. Malignancy is heterogeneous, and the prognosis varies even in patients with the same pathological stage. While some patients overcame the disease, others suffered from local recurrence and distant metastasis. Some aspects of the disease cannot be explained by the TNM staging system, but the exposition of molecular makers can help uncover the mechanisms and predict the outcomes. Molecular therapy has become an inevitable trend with the development of precision medicine, and the novel model of EAGs is more effective in BRCA patients with gene mutations. Compared with traditional treatment, BRCA patients can be classified through a risk score model to determine the greater or less urgency of therapy. Hence, the prognostic risk score model could support personalized therapeutic strategies in clinical practice and control recurrence and distant metastasis in BRCA.
Despite the limitations of retrospective research, our EAGs signature was validated in TCGA integral dataset and UCSC and GEO external cohorts. Since our EAGs signature satisfied the consequences, even in mixed cohorts involving different types of BRCA, we believe that the results from EAGs signatures are reliable. Most genes in this model have been previously reported to be involved in cancer. For example, chemokine ligand 13 (CXCL13) and its chemokine receptor 5 (CXCR5) form the CXCL13/CXCR5 signaling axis to modulate the ability of cancer cells to grow, proliferate, invade, and metastasize. Moreover, the axis was able to play a role in the microenvironment to promote immune escape in cancers. 22 Overexpression of VWCE inhibits the invasion and metastasis of BRCA lines by downregulating the expression of WDR1, which acts as a downstream molecule, indicating that VWCE may represent a novel tumor suppressor. 19 KDM4B has also been reported to promote glucose uptake and ATP production by regulating the expression of GLUTs and AKT signaling pathways in colorectal cancer. In summary, there is a correlation between KDM4B and glucose metabolism. 23 Moreover, HGF could form a signaling axis, enhancing proliferation, invasion, and metastasis in head and neck squamous cell carcinoma. In addition, HGF in the tumor microenvironment assists immune surveillance and immune activation. 24 As has been acknowledged that MMP9 mainly exists in extracellular matrix, resolving the adhesion of cell-to-cell. Recently, an increasing number of researchers have explored the signal-mediated function of MMP9 in the microenvironment. 25 PAK1 was shown to induce resistance to hypoxia by regulating hypoxia-inducible factors in pancreatic cancer. 26 Maier et al 27 revealed that LEF1 may be related to tumoral antigen presentation, and activating the Wnt/β-catenin signaling in adrenocortical carcinoma. CCL5 and CCL19 have both been identified as chemokines based on their ability to induce chemotaxis. They can combine with their ligands, which have a great effect on inflammation and immune surveillance in cancer progression. 28 The SIAH2/HIF-1 axis triggered by low oxygen tension can remodel the tumor microenvironment by regulating tumor mitochondrial function, resulting in the enhancement of the Warburg effect, metabolic reprogramming, and tumor immune response. 29 TNFSF11 is known as RANKL, and TNFRSF11A (also called RANK) is considered the sole receptor for RANKL. Their primary role is in the regulation of bone remodeling and the development of the immune system. 30 Research has suggested that RANKL inhibition might delay bone metastasis or disease recurrence in patients with early-stage BRCA. 31 The first evidence of CPEB1 involvement in gastric cancer is presented, companying with the molecular mechanism underlying the regulation of its expression and its potential role in invasion and angiogenesis. 32 Carly et al 33 found that ACKR4 can inhibit CD103+ dendritic cell retention in tumors by regulating the intratumor abundance of CCL21, and the behavior of intratumor T-cell accumulation and activation can alter tumor growth. Furthermore, the PROK/PROKR system has been associated with a considerable number of physiological and pathological functions, particularly potent angiogenic and immunoregulatory activities in different types of cancer. 34 Based on several research annotations for these 15 genes, we can conclude that the process of EMT correlates with metabolic reprogramming, the tumor immune microenvironment, angiogenesis, CSC properties, and extracellular chemokines.
In summary, beyond individual associations, the collective function of these genes reveals a coherent biological narrative centered on EMT. They operate as an integrated network in key processes that drive malignancy, CXCL13, CCL5, and CCL19 modulate immune and cytokine signaling within the tumor microenvironment, PAK1 regulates cytoskeletal dynamics and cell motility, MMP9 facilitates extracellular matrix degradation, while KDM4B and SIAH2 influence metabolic and epigenetic reprogramming. It is the concerted dysregulation of this network rather than any single gene that appears to define the aggressive phenotype associated with our model. This systems-level perspective suggests that our signature reflects interconnected biological pathways associated with EMT.
The advantage of this study is that we identified 15 EAGs as a new prognostic signature to predict survival time in BRCA. The relatively high area under the curve and nearly coincident calibration curve in the validation cohort enhanced the reliability of the model. In addition to the potential clinical significance of our study, several limitations must be considered. First, the clinical parameters from TCGA, UCSC, and GEO databases are limited and incomplete, and potential factors such as personal history, background diseases, chemotherapy, radiotherapy, and targeted drug therapy are missing in the research, which may affect the identification of these gene signatures. Second, the modest predictive accuracy of our signature is a key limitation, it clearly positions the model as a complementary tool, not a standalone diagnostic, and future improvements hinge on deeper clinical and molecular data. Third, the IHC validation was based on public database images. While this provided potential supportive evidence, future studies employing more standardized tissue microarrays and more precise quantitative scoring were needed to confirm the protein level expression differences of these EMT signature genes. Finally, the function of these genes in EMT require direct experimental validation in vitro and in vivo, and independent prospective trials are necessary to confirm the prognostic risk-score model, and the value of these genes as potential pharmacological targets requires further investigation.
Conclusions
In summary, this study developed and validated a 15-gene prognostic signature based on EMT for BRCA through integrative bioinformatics analysis of multiple public cohorts. The signature demonstrated the independent prognostic value for patient survival and showed association with metastatic potential, underscoring the relevance of EMT biology in disease progression. Furthermore, the integration of this signature with key clinical parameters into a nomogram provided a practical tool for individualized risk assessment. Future prospective studies in independent clinical cohorts and functional investigations of the identified genes were warranted to confirm the clinical utility of this model and to elucidate the underlying biological mechanisms.
Supplemental Material
sj-docx-1-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-docx-1-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-docx-2-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-docx-2-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-docx-3-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-docx-3-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-4-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-tif-4-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-5-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-tif-5-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-6-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-tif-6-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-7-bcb-10.1177_11782234261433697 – Supplemental material for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis
Supplemental material, sj-tif-7-bcb-10.1177_11782234261433697 for Genes From Epithelial-Mesenchymal Transition Predict Overall Survival Effectively in Breast Cancer: A Novel Risk Model Based on Initial Step of Tumor Metastasis by Wei Liang, Zi-Ying Wang, Quan-Feng Shao, Yuan-Yuan Li, Bei Zhu, Xi-Hu Qin and Wei-Xian Chen in Breast Cancer: Basic and Clinical Research
Footnotes
Acknowledgements
The authors thank Dr Qi Qian, Dr Jian-bo Dai, and Dr Shu-yang Xu for providing technical assistance.
Ethical Considerations
Ethics approval is not required.
Consent to Participate
N/A.
Consent to Publish
N/A.
Author Contributions
Wei Liang: Conceptualization, Data curation, Writing – original draft.
Zi-ying Wang: Data curation, Formal analysis, Resources, Writing – review & editing.
Quan-feng Shao: Data curation, Investigation, Validation.
Yuan-yuan Li: Investigation, Resources, Software, Visualization.
Bei Zhu: Data curation, Validation, Visualization.
Xi-hu Qin: Conceptualization, Software, Supervision, Validation.
Wei-xian Chen: Conceptualization, Writing – review & editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was kindly supported by grants from the Changzhou Medical Center (CMCB202401), the Jiangsu Provincial Health Commission General Project (MQ2024036), the Excellent Post-doctoral Program of Jiangsu Province (2022ZB820), the Top Talent of Changzhou “The 14th Five-Year Plan” High-Level Health Talents Training Project (2022CZBJ065), and the Post-doctoral Foundation of China (2022M720543).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets including patients’ personal clinical information are available in the TCGA (https://portal.gdc.cancer.gov/), UCSC (https://ucscpublic.xenahubs.net), and GEO database (![]() ). The custom R code used to perform the core analyses, including differential expression, risk modeling, and nomogram construction, is available from the corresponding author upon reasonable request for the purpose of replicating the findings.
). The custom R code used to perform the core analyses, including differential expression, risk modeling, and nomogram construction, is available from the corresponding author upon reasonable request for the purpose of replicating the findings.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.