
Abstract
Background:
Ferroptosis is a recently studied form of programmed cell death characterized by lipid peroxides accumulation in the cells. This process occurs when a cell’s antioxidant capacity is disturbed resulting in the inability of the cell to detoxify the toxic peroxides. Two major components that regulate ferroptosis are cysteine and iron.
Objective:
This study aimed to determine the effect of cysteine deficiency and iron chelation on triple-negative breast cancer (TNBC) ferroptosis in a lipid-enriched microenvironment.
Design:
The study has a laboratory-based experimental design. This study used the MDA-MB-231 cell line in various in vitro cell culture systems to investigate the research question.
Methods:
For the first part of the study, we subjected MDA-MB-231 cells to grow in cysteine-absent adipocyte-conditioned media. In the second half, we treated MDA-MB-231 cells with iron chelator, deferoxamine. BODIPY imaging and western blot were carried out to observe ferroptosis in the cells under the 2 conditions.
Results:
The results showed that cysteine absence in the conditioned media was able to reduce the formation of lipid droplets, which increased the greater access to free fatty acids to undergo oxidation, therefore inducing ferroptosis. On the contrary, cells when treated with deferoxamine along with erastin (ferroptosis-inducing drug), showed an increase in cell iron content was observed, later inducing ferroptosis.
Conclusion:
Our results show an alternative function of cysteine and deferoxamine, one regulating lipid droplets and the other inducing ferroptosis, although an inhibitor of the same, respectively.
Plain Language Summary
The lack of this nonessential amino acid was however able to sensitize triple-negative breast cancer cells to ferroptosis, even in the presence of adipocytes. Iron chelator such as deferoxamine was also affected by the absence of cysteine, wherein they aggravated the situation by increasing the intracellular iron content, therefore promoting ferroptosis. Treatment with System xc inhibitor (erastin or sulfasalazine) and deferoxamine showed similar results.
Introduction
Programmed cell death in cancer has emerged as an alternative force for eradicating cancer cells. Recent studies are focusing on cell death of malignant tumors, which also includes ferroptosis (an iron-based cell death). Ferroptosis is driven by the accumulation of lipid peroxides within the cells. 1 Its initiation in mammalian cells mainly depends on iron, lipid, and glutathione-dependent redox equilibrium. 2 Induction of ferroptosis and inhibition of tumor growth have been reported across multiple cancer types (lung, breast colorectal, liver, gastric, and melanoma). 3
In breast cancer, abnormal metabolism of amino acids and lipids has been observed, which are strongly associated with ferroptosis. 4 Of particular interest is the potential role of ferroptosis in triple-negative breast cancer (TNBC), a subtype of breast cancer characterized by estrogen receptor (ER) and progesterone receptor (PR) expression and no amplification in HER2. 5 This class of tumor has worse overall survival, and targeted therapeutic options are limited; hence, there is a need for continuous research to identify newer therapeutic options. 6 Compared to the other types of breast cancer, TNBC cells have shown to be more sensitive to ferroptosis, either by inducing cysteine inhibition or inhibiting glutathione peroxidase-4 (GPX4), a lipid peroxide neutralizing enzyme.7,8 Targeted induction of ferroptosis may hold potential as an alternative modality to treating TNBC. 9
Cysteine and iron are 2 important factors regulating ferroptosis, the former inhibiting it and the latter inducing it. 10 Cysteine is important for the synthesis of glutathione (GSH) which in turn is important for the activity of GPX4, 11 thereby enhancing cellular detoxification from drugs.12,13 This activity confirms the role played by cysteine in the growth, development and treatment resistance in tumors. 14 Iron, on the contrary, is required for the oxidation of polyunsaturated fatty acids (PUFAs) to lipid peroxides.15,16 For the prognosis of TNBC, studies have shown the importance of iron, suggesting a potential for treatment with iron chelators for a highly metastatic subpopulation of TNBCs.17,18
Cysteine is an important amino acid required for the development of TNBC, inducing ferroptosis during its deficiency. 19 System xc is the famous cellular cysteine transporter in cells. 1 Inhibition of this transporter by Sulfasalazine or Erastin has been shown to slow down the metabolism, GSH level depletion and reactive oxygen species (ROS) amplification, therefore leading to ferroptosis in TNBCs.20,21
Breast cancer grows in a high-adipocytic environment unique from most cancers. Factors released from adipocytes have shown potential in enhancing the growth and proliferation of breast cancer. 22 A recent study has shown that mammary adipocytes can inhibit ferroptosis in breast cancer cells, suggesting a potential difficulty in enhancing ferroptosis in clinical studies via ferroptosis. 23
The overall poor survival and lack of targeted therapy modalities in TNBC combined with the potential for adipocytes to inhibit ferroptosis, creates the opportunity to explore alternative methods to induce ferroptosis in breast cancer. Synergistic effects between chemotherapeutic drugs and ferroptosis-inducing drugs have shown positive results in TNBC treatment. 24
In this study, we explored the effect of adipocyte-conditioned media in regulating ferroptosis in TNBC, in the presence or absence of cysteine along with the regulating effect played by deferoxamine (DFO). Our study suggests a new therapeutic approach for using DFO and ferroptosis-inducing drugs to overcome TNBC.
Materials and Methods
Antibodies and reagents
Antibodies used are as follows: anti-GPX4 (ab125066, abcam), anti-perilipin2 (NB110-40877, novusbio), GAPDH (GTX100118, Genetex), anti-transferrin receptor (A22161, ABclonal), and anti-ACSL4 (ab155282, Abcam). Deferoxamine (D9533) and sulfasalazine (S0883) were purchased from Sigma. Erastin (S7242), Ferrostatin-1 (Ferr-1) (S7243), and RSL3 (S8155) were purchased from Selleckchem. Ferro-orange dye (F374) was purchased from Dojindo and BODIPY C11 (D3861) and LD BODIPY and Mitotracker (M7512) were purchased from thermofisher Invitrogen.
Cell culture
We purchased a human breast cancer cell (MDA_MB_231) from Korea Cell Line Bank (South Korea). RPMI 1640 (LM011-03, Welgene) containing 10% fetal bovine serum (16000044, Gibco) and 1% penicillin/streptomycin (LS202-02, Welgene) was used to culture the cells at 37°C with 5% CO2. MCF10A, a nontumorigenic breast epithelial cell was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% horse serum and 1% penicillin-streptomycin.
Adipocyte culture and isolation of ADCM and cdAM
Adipose tissue-derived mesenchymal stem cells (CEFO-ADMSC-kit) were purchased from CEFO (cell engineering for origin). Primary adipocytes were expanded in DMEM (Welgene) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Welgene). Adipocyte cells were differentiated, and the conditioned media was collected according to the process described by Gyamfi et al. 22
For obtaining cysteine-free ADCM (cdAM), after the maintaining process, cells were incubated in cysteine-free media for 48 hr and after the incubation period, the media was collected.
Cell viability
Cell counting kit-8 (also known as CCK-8) (CCK-3000, Dongindo) was used to assess cell viability. In brief, the cells were seeded in 96-well plates and incubated with the respective treatments. Following the incubation period, cells were incubated with 10 ul of CCK-8 reagent, according to the protocol provided by the supplier. After incubating for 2 hours, an absorbance value at 450 nm was detected (Tecan Group Limited, Mannedorf, Switzerland).
Hematoxylin and eosin staining
20x104 cells were seeded in a 6-well plate. In the following days, cells were treated with respective drugs or vehicles in a serum-free medium and incubated at 37°C for 48 hours. After the incubation period, cells were fixed with 4% paraformaldehyde and stained with hematoxylin for 5 mins followed by eosin for 5 mins. Cells were washed with distilled water and viewed under Olympus BX53 microscope (Olympus Optical Co., Tokyo, Japan). The length of the cells was calculated using ImageJ software.
RNA extraction and quantitative real-time PCR
Total RNA from cells cultured in ADCM/cdAM, with/ without DFO for 24 hr and 48 hr was extracted using the RNeasy Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s guidelines. For rtPCR, TOPreal One-step RT-qPCR kit (SYBR Green with high ROX, enzynomics, Korea) was used with 50 ng of RNA and analyzed using the StepOne Plus Real-time PCR system (Applied Biosystems, Foster City, CA, USA). All the reactions were performed in triplets, with GAPDH as the housekeeping gene. Relative gene expression was evaluated by comparative CT method (2-ΔΔCT). The primers used are listed in the Supplementary Table 1.
Protein extraction and western blot
Cells were lysed in RIPA buffer (GenDEPOT, TX, USA) containing 1% protease inhibitor cocktail (GenDEPOT). 15ug of each sample was separated using SDS-PAGE and was then transferred to 0.45um nitrocellulose membrane. Western blot was performed as previously described by Liu et al, 25 with the antibodies listed above. The protein bands were visualized using enhanced chemiluminescence reagents (Western Lighting Plus, PerkinElmer, USA).
BODIPY staining and confocal imaging
In cell culture slide (30108, SPL) 2x104 cells were seeded and cultured with either ADCM/ cdAM or respective drugs for 24 hours and 48 hours. After the incubation period, the cells were washed with phosphate buffered saline (PBS), fixed and stained with either BODIPY C11 (1.5 uM), Ferro-orange (1 uM), or LD BODIPY (2 uM) for 30 minutes in PBS. After staining, the cells were washed with PBS, and the nucleus was stained with DAPI for 5 minutes. After washing the cells with PBS, the cells were immediately viewed under ZEISS LSM 710 microscope (ZEISS, Germany).
Statistical analysis
For analyzing the statistical significance of the experiments, GraphPad Prism version 5 (GraphPad Software, San Diego, CA, USA) was used. Results are shown as the means, and the error bars represent the standard error of the means (SD). Two or more independent experiments were performed. The statistical significance was determined by unpaired Student’s t-test. P values <.05 were considered to show a significant difference statistically.
Results
Cysteine absent adipocyte conditioned media sensitizes TNBC cell to ferroptosis
To examine the effect of adipocyte conditioned media (ADCM) on ferroptosis, we performed cell viability using CCK assay with or without a ferroptosis-inducing agent RSL3. The TNBC cell line (MDA-MB-231) was cultured in the presence or absence of ADCM and was treated with RSL3. Cell counting kit assay results showed a significant difference between cells treated with and without RSL3 (P = .0294; P ⩽ .0001). MDA-MB-231 cells which are highly sensitive to ferroptosis, had lost their sensitivity when culture with ADCM (Figure 1A).

Induction of ferroptosis by adipocyte-conditioned media in the absence of cysteine. (A-C) Cell viability of MDA-MB-231 cells under the mentioned conditions using CCK8 assay, which showed the decrease in cell viability after treatment with cdAM. (D) BODIPY staining of MDA-MB-231 with BODIPY C11, after culturing in ADCM or cdAM; showed higher accumulation of oxidized lipids when cultured in cdAM. (E) Western blot images for GPX4, ACSL4, and TFR1 of MDA-MB-231 cells cultured in ADCM or cdAM, confirming the induction of ferroptosis in cdAM-cultured cells. Each experiment was performed in 3 replicates; unpaired t-test statistical analysis was performed for CCK assay. (Data indicate mean ± SEM; ***P < .001; **P < .01; *P < .05).
Cysteine is a major requirement for the inhibition of ferroptosis, and its absence can do the opposite. To determine if cysteine plays a role in ferroptosis regulation in TNBC, TNBC cells were cultured in cysteine-free media and treated with or without RSL3. Cell viability results showed that in the absence of cysteine, cells were more sensitive to ferroptosis in the presence of RSL3, compared with control cells (Figure 1B) (P = .0001).
Based on the above results, we eliminated cysteine from ADCM (cdAM (cysteine-depleted adipocyte media)). This was carried out to determine if cdAM exerts a similar effect to ADCM in regulating ferroptosis. Cell counting kit assay results showed that cdAM restored the sensitivity of MDA-MB-231 cells to ferroptosis (Figure 1C; P = .0001). To confirm that the cell death caused by cdAM was ferroptosis, we obtained BODIPY images, wherein the cells were stained with BODIPY C11 dye. The images showed an increase in the intensity of the dye in cells cultured in cdAM compared with ADCM, confirming ferroptosis occurrence (Figure 1D). We subsequently performed a western blot to determine the protein expression of ferroptosis-related genes (GPX4, ACSL4, and Transferrin receptor 1). Blot images showed a decrease in the expression of GPX4, along with an increase in the expression of TFR1 and ACSL4 (Figure 1E).
These results confirm that the absence of cysteine in ADCM can change the latter’s effect from desensitizing to ferroptosis sensitizing.
Cysteine-depleted adipocyte media mediate ferroptosis by reducing the formation of lipid droplets and increasing the availability of free fatty acids
To understand the mechanism behind cdAM-induced ferroptosis, we first examined the adipocyte cells from which cdAM were collected. The morphological feature showed lipolytic-like characteristics in the lipid droplets (LDs) of the adipocyte cells maintained in cysteine-free media (Figure 2A). We hypothesized that LD lipolysis is potentially the mechanism behind cdAM-induced ferroptosis. To confirm this theory, MDA-MB-231 cells were seeded in cdAM or ADCM for 24 and 48 hours, and LDs were examined by staining the cells with LD BODIPY. BODIPY images showed that ADCM noticeably led to LD formation in the cells, which insignificantly increased with time. In cdAM cultured cells, although the cells showed the presence of LDs, their amount reduced with the increase in culture time (Figure 2B). This observation suggests that cysteine absence can affect the LD formation within the cells, allowing for the availability of free fatty acids to undergo lipid peroxidation.
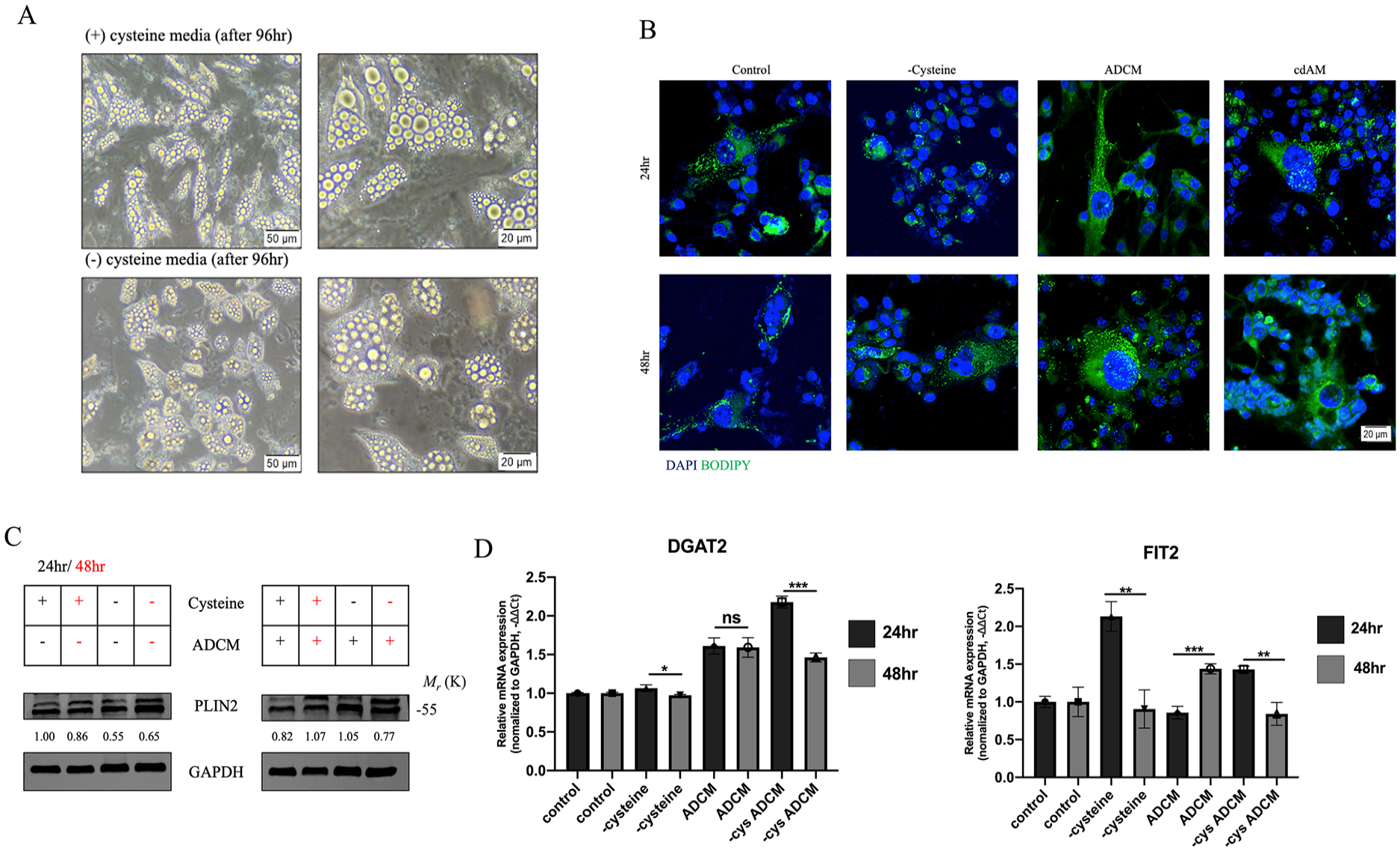
cdAM reduces the formation of lipid droplets, which sensitizes cells to ferroptosis. (A) Morphological characteristics of adipocyte cells when maintained in the presence or absence of cysteine in the media; disfigured lipid droplets were observed in adipocytes cultured without cysteine. (B) BODIPY staining of MDA-MB-231 cells with LD BODIPY after culturing in ADCM or cdAM; lower accumulation of lipid droplets in cdAM cultured cells was observed. (C) Western blot image of PLIN2 after culturing the cells in ADCM or cdAM for 24 and 48 hours; displayed a reduction in the expression of PLIN2 in cells cultured in cdAM. (D) Quantitative PCR (qPCR) comparing the expression of lipid droplet formation genes (DGAT2 and FIT2) in MDA-MB-231 cells after culturing in ADCM and cdAM media; presented a decrease in the expression of the 2 genes in cdAM condition. Relative mRNA expression was normalized to GAPDH. Each experiment was performed in 3 replicates; unpaired t-test statistical analysis was performed for RTq-PCR. (Data indicate mean ± SEM; ***P < .001; **P < .01; *P < .05).
We further measured the expression of some important genes which are implicated in LD formation. As per the western blot images, the protein expression of PLIN2 (perilipin 2) was higher in ADCM-cultured cells compared with cdAM (Figure 2C). Similar expression pattern was observed with the mRNA expression of DGAT2 (Diacylglycerol O-Acyltransferase 2) (P = .0001) and FIT2 (Fat Storage Inducing Transmembrane Protein 2) (P = .0029; Figure 2D). These results therefore gave away a confirmatory explanation for the LD images.
The above data propose that cdAM induces ferroptosis in cells by reducing LD formation within the cells, which therefore makes them vulnerable to ferroptosis.
DFO induces ferroptosis in cysteine deprivation conditions
Iron is a major component for the induction of ferroptosis, as it is responsible for the production of lipid peroxides, and its depletion or chelation will result in the opposite. To investigate the effect of iron chelation in our experimental system, we treated the cells with DFO to observe ferroptosis inhibition. Iron chelation by DFO caused a significant increase in the expression of transferrin receptor (P = .0029) and a decrease in ferritin heavy chain (P = .0029) in cdAM cultured cells; however, the same was not observed in ADCM-cultured cells (Figure 3A).

Iron chelation by deferoxamine induces ferroptosis in the absence of cysteine. (A) Quantitative PCR (qPCR) comparing the expression of iron-regulating genes (ferritin and transferrin) after culturing the cells in ADCM or cdAM media; the results depicted the decrease of ferritin with an increase in transferrin in cdAM cells treated with DFO. Relative mRNA expression was normalized to GAPDH. (B) BODIPY staining of MDA-MB-231 cells with BODIPY C11, cultured in ADCM or cdAM, in the presence or absence of DFO; cells were able to show oxidized lipid accumulation, when cultured in cdAM condition along with DFO. (C) Western blot images of GPX4, ACSL4, and TFR1, after culturing the cells in ADCM or cdAM, with or without DFO; presented a confirmatory ferroptosis results for the above presented confocal results (D-E) Hematoxylin and eosin staining, and cell length quantification of cells treated with DFO and Ferrostatin-1; unfolded the cell death type behind DFO treatment. Each experiment was performed in 3 replicates; unpaired t-test statistical analysis was performed for RTqPCR. (Data indicate mean ± SEM; ***P < .001; **P < .01; *P < .05).
While treating the cells with DFO, we observed that long-duration DFO treatment led to morphological changes in the cells.
Studies have suggested that DFO treatment can lead to autophagy cell death, hence, we treated MDA-MB-231 cells with DFO and chloroquine (CQ), an autophagy inhibitor to understand if morphological changes observed may be autophagy. Morphological changes were observed after 48 hours of treatment; however, CQ was unable to reverse the action of DFO, suggesting a type of cell death apart from autophagy (Supplementary Figure 1A). To confirm if the DFO effect is a cancer cell-specific event, we treated MCF10A (normal breast cancer cells) with DFO for 48 hours. In MCF10A cells, there were no changes in the morphology of the cells (Supplementary Figure 1B-C).
Since DFO-induced cell death showed not to be autophagy, we went on to confirm if the cell death type was ferroptosis. BODIPY images by staining DFO treated/nontreated cells with BODIPY C11 were captured. Deferoxamine-treated cdAM cells showed an increase in the intensity of the dye when compared with ADCM cells (Figure 3B), suggesting that longer exposure to DFO can still induce ferroptosis-like cell death. Western blot images also showed an increase in the expression of ACSL4 and TFR1 in these cells, further confirming the data (Figure 3C). To further confirm our theory, MDA-MB-231 cells were treated with DFO along with different concentrations of ferrostatin-1, another type of ferroptosis inhibitor. With the increase in the concentration of Ferr-1, the cells regained their original morphology (Figure 3D and E). These results suggest that DFO can induce ferroptosis when treated for a longer duration, specifically in cysteine-absent conditions.
DFO increases the intercellular iron content in the absence of cysteine which induces ferroptosis
Our observation of DFO-induced ferroptosis in cysteine-absent and cdAM cells led us to explore the mechanism behind this. Experiments presented from hereon did not use cdAM and ADCM. Since the absence of cysteine played a major role in DFO-induced ferroptosis, the cells were treated with erastin (SLC7A11 inhibitor; ferroptosis inducer). When treated with erastin along with DFO, an increase in the intracellular iron was observed in the cells when compared with control samples after 48 hours treatment (Figure 4A). Intracellular iron level was analyzed by staining the cells with ferro-orange, an iron staining dye. Since iron is required for ferroptosis, BODIPY C11 imaging was carried out for these cells. A 48-hour treatment duration showed an increase in the intensity of the dye compared with 24-hour treatment set (Figure 4B). To further confirm the confocal imaging results, protein expression of ferroptosis-related genes also showed induction of ferroptosis (Figure 4C). The above results suggest that although DFO is supposed to inhibit ferroptosis, it has an alternative effect in the absence of cysteine.

Erastin and deferoxamine induce ferroptosis in TNBC by increasing intracellular iron content. (A) Confocal imaging of MDA-MB-231 stained with ferro-orange, after treatment with erastin and deferoxamine for 24 and 48 hr; increased intracellular iron after treatment with erastin and DFO was visualized. (B) BODIPY staining of MDA-MB-231 with BODIPY C11 after treatment with deferoxamine and erastin for 24 and 48 hr; increase in oxidized lipids was observed under combined drug treatment. (C) Western blot images of ferroptosis-related genes after the indicated treatments; presented and confirmed the induction of ferroptosis in the presence of erastin and DFO. (D) Expression of the transferrin receptor in BRCA TCGA sample and overall survival. Each experiment was performed in 3 replicates.
Using the Broad et al. dataset from cBioPortal, we performed bioinformatic analysis to check the expression of transferrin receptors in BRCA (Breast Cancer gene) patients. 26 A similar expression pattern of TFR1 was observed in our results as well (Figure 4C and D). However, DFO and erastin treatment were able to reverse it by increasing the expression, suggesting a therapeutic approach.
We further confirmed the above results with another ferroptosis-inducing drug, sulfasalazine (SAS), and the results were similar to erastin, therefore providing an alternative for erastin in clinical approaches (Supplementary Figure 2A-C). Ferroptosis is shown to dysregulate mitochondria morphology by reducing cristae, mitochondrial membrane density increases, and a decrease in mitochondrial volume.27,28 To see if the intensity of mitochondrial expression reduces in these cells, confocal imaging was performed by dying the cells with a mitotracker. The results clearly showed a decrease in the intensity of the dye, therefore suggesting the changes caused by ferroptosis on mitochondria (Supplementary Figure 2D).
Discussion
Ferroptosis, although a relatively novel form of programmed cell death, has rapidly gained prominence due to its potential therapeutic applications, particularly in cancer treatment.29,30 Our study builds on existing literature by revealing alternative functions of cysteine and DFO in regulating ferroptosis in TNBC cells.21,6 Unlike conventional perspectives,31,13 our findings suggest that both cysteine depletion and iron chelation by DFO act synergistically to sensitize TNBC cells to ferroptosis under specific conditions.
Cysteine, typically considered a nonessential amino acid, assumes a critical role in cellular physiology due to its involvement in LD formation and redox balance. Prior research has highlighted cysteine’s relationship with LDs through imaging probes that target these structures. 32 Our study extends these observations by demonstrating that cysteine depletion disrupts LD accumulation, thereby increasing the availability of free PUFAs for peroxidation (Figure 2). This disruption sensitizes cells to ferroptosis, aligning with studies that identify LDs as protective reservoirs against PUFA oxidation.33-35 Although in vivo cysteine depletion poses challenges, the development of cyst(e)inase—a synthetic enzyme designed to deplete cysteine and cystine—offers a promising avenue for clinical translation.36,37
Interestingly, our findings show that DFO, traditionally known as an iron chelator that inhibits ferroptosis, 38 exhibits an unexpected pro-ferroptotic effect in cysteine-depleted conditions. Deferoxamine increases intracellular iron levels, which facilitates lipid peroxidation and ferroptosis. This observation underscores the context-dependent effects of DFO, particularly in TNBC cells, which are inherently more sensitive to ferroptosis. This dual functionality of DFO aligns with previous studies suggesting its potential as an anti-cancer agent 39 (https://clinicaltrials.gov/study/NCT05300958), but our findings provide new insights into its mechanisms, particularly under cysteine-deficient conditions.
The observed interplay between cysteine depletion and DFO treatment also highlights the critical role of ferritin regulation in ferroptosis. Our results demonstrate a downregulation of ferritin in cysteine-depleted cells, further promoting free iron availability and enhancing ferroptosis (Figure 3A). These findings align with research showing that ferritin knockdown sensitizes cancer cells to treatments by disrupting LD stability. 40
Despite its therapeutic potential, DFO’s clinical applicability is limited by poor oral bioavailability, necessitating alternative delivery strategies.41,42 Synthetic iron chelators are emerging as promising substitutes, potentially improving the efficacy and safety of ferroptosis-based therapies. 43 Moreover, combining cyst(e)inase with ferroptosis-inducing agents like DFO could amplify the therapeutic effects, 36 although further studies are needed to optimize this combination for both in vitro and in vivo applications.
In conclusion, our study reveals a novel mechanism by which cysteine depletion and DFO synergistically induce ferroptosis in TNBC cells. By disrupting LD physiology and increasing intracellular iron levels (Figure 5), this combinatory approach leverages the vulnerabilities of TNBC cells to ferroptosis. These findings pave the way for innovative therapeutic strategies targeting aggressive tumor types through ferroptosis induction. Further investigations are warranted to validate these findings in clinical settings and to explore the feasibility of implementing cyst(e)inase and iron chelators in cancer therapy.

Schematic representation on how absence of cysteine and presence of iron chelator (deferoxamine) work together to disrupt the physiology of lipid droplets and increase the intracellular iron content in cells, respectively. This co-working system increases the oxidation of lipids, therefore initiating ferroptosis.
Conclusion
The study presents the importance of cysteine in maintaining the physiology of LDs, whose absence has been shown to affect the latter’s accumulation within cells. We show that the lack of this nonessential amino acid was however able to sensitize TNBC cells to ferroptosis, even in the presence of adipocytes. Iron chelator such as DFO was also affected by the absence of cysteine, wherein they aggravated the situation by increasing the intracellular iron content, therefore promoting ferroptosis. Treatment with System xc inhibitor (erastin or sulfasalazine) and DFO showed similar results. In conclusion, drug combination therapy between a ferroptosis-inducing drug (specifically a cysteine inhibitor drug) and an iron chelator can pose a beneficial alternative for treating aggressive tumor types, such as TNBC.
Supplemental Material
sj-docx-1-bcb-10.1177_11782234241311012 – Supplemental material for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells
Supplemental material, sj-docx-1-bcb-10.1177_11782234241311012 for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells by Manvi Agarwal Neeraj and JunJeong Choi in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-docx-2-bcb-10.1177_11782234241311012 – Supplemental material for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells
Supplemental material, sj-docx-2-bcb-10.1177_11782234241311012 for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells by Manvi Agarwal Neeraj and JunJeong Choi in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-docx-3-bcb-10.1177_11782234241311012 – Supplemental material for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells
Supplemental material, sj-docx-3-bcb-10.1177_11782234241311012 for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells by Manvi Agarwal Neeraj and JunJeong Choi in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-4-bcb-10.1177_11782234241311012 – Supplemental material for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells
Supplemental material, sj-tif-4-bcb-10.1177_11782234241311012 for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells by Manvi Agarwal Neeraj and JunJeong Choi in Breast Cancer: Basic and Clinical Research
Supplemental Material
sj-tif-5-bcb-10.1177_11782234241311012 – Supplemental material for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells
Supplemental material, sj-tif-5-bcb-10.1177_11782234241311012 for Absence of Cysteine and Iron Chelation Induces Ferroptosis in Triple-Negative Breast Cancer Cells by Manvi Agarwal Neeraj and JunJeong Choi in Breast Cancer: Basic and Clinical Research
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.