
Abstract
Histone deacetylases (HDACs) are essential epigenetic regulators, with HDAC6 overexpression linked to estrogen receptor (ER) activity and breast cancer progression. While several HDAC6 inhibitors have been investigated, their clinical success remains limited due to toxicity and off-target effects, necessitating the discovery of novel, selective inhibitors. This study employs a multi-stage computational approach to identify potent HDAC6 inhibitors for breast cancer therapy. A large-scale virtual screening of 264 834 compounds was conducted, followed by molecular docking, molecular dynamics (MD) simulations (100 ns), molecular mechanics/generalized born surface area (MM/GBSA) binding free energy calculations, and absorption, distribution, metabolism, excretion, and toxicity (ADMET) predictions. The HDI-3 emerged as the most promising candidate among replicate simulations, exhibiting a substantially favorable MM/GBSA binding free energy of −130.67 kcal/mol—indicative of strong thermodynamic stability and stronger binding affinity compared to reference inhibitors Trichostatin A and Ricolinostat. Molecular dynamics simulations revealed that HDI-3 maintained structural stability, persistent key interactions with active site residues (ASP649, HIS651, ASP742), and low conformational fluctuations. The ADMET evaluation confirmed HDI-3’s favorable pharmacokinetic properties, including optimal bioavailability, non-mutagenicity, and low hepatotoxicity. Essential dynamics and principal component analysis further validated its stable binding profile. While these findings highlight HDI-3 as a selective and pharmacologically viable HDAC6 inhibitor, it is important to acknowledge that the results are entirely computational. Therefore, experimental validation is essential to confirm the compound’s efficacy and safety. This integrated computational pipeline provides an efficient strategy to accelerate targeted drug discovery, laying the groundwork for future experimental investigations.
Introduction
Breast cancer is one of the most common tumors worldwide, with approximately 2.2 million new breast cancer cases and 684 996 deaths reported in 2020.1,2 Breast cancer is considered the most prevalent cancer type and is estimated to account for 30% of all new cancers detected in women, with a mortality rate of 6% worldwide. 2 Breast cancer 3 is a complex and heterogeneous disease characterized by abnormal cell proliferation of the epithelial cells lining the milk ducts. 4 Breast cancer is a highly diverse disease characterized by variations in cancer cells’ epigenomic, genomic, proteomic, and transcriptomic profiles. 5 Epigenetic modifications, such as histone modifications and DNA methylation, have been identified as key contributors to breast cancer development.6 -8 Histone modification is a key epigenetic mechanism that includes acetylation, methylation, and phosphorylation.6,9 Histone acetylation plays a crucial role by targeting the acetyl groups on lysine residues, promoting chromatin relaxation and transcriptional activation. 10 This process is regulated by 2 enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). The HATs facilitate transcription by acetylating lysine residues on histone tails, maintaining an open and accessible chromatin structure. In contrast, HDACs remove acetyl groups, leading to chromatin condensation and gene repression. An imbalance between the activities of HATs and HDACs can disrupt gene expression by altering DNA accessibility, contributing to cancer development. 10
There are 18 HDAC isoforms in humans, which are categorized into 4 different classes. 11 Class I (HDAC1, HDAC2, HDAC3 and HDAC8), Class IIa (HDAC4, HDAC5, HDAC7 and HDAC9), Class IIb (HDAC6 and HDAC10), and class IV (HDAC11) are Zn+2-dependent metalloenzymes. Class III HDAC belongs to sirtuins (SIRT1-SIRT7), which are dependent on nicotinamide adenine dinucleotide (NAD+). 11 Recent studies have highlighted that Zn+2-dependent HDACs are overexpressed in various tumors like breast, prostate, and liver cancers, multiple myeloma, and neuroblastoma, especially class I and class II HDAC isoforms. 12
The HDAC6 is distinct from other histone deacetylases due to its cytoplasmic localization and its activity on multiple non-histone proteins, including surviving, cortactin, α-tubulin, Hsp90, heat shock factor (HSF-1), ERK1, Ku-70, Micro-1, and peroxiredoxin. 13 Among these, acetylation of α-tubulin at lysine 40 was the first identified substrate and remains one of the extensively studied. 13 Structurally, HDAC6 consists of 2 catalytic domains (CD1 and CD2), which provide the enzymatic core required for its deacetylase activity. 14 Because of this unique architecture and its broad range of substrates, HDAC6 has attracted significant attention for its role in numerous biological and pathological processes. The HDAC6 links extensive evidence to the regulation of cancer progression, neurodegenerative disorders, and immune-related diseases. 15 In cancer, HDAC6 overexpression promotes cytoskeletal reorganization, enhances cell motility, facilitates endothelial migration, and triggers both angiogenesis and metastasis. 16 The HDAC6 expression has been regulated by estrogen signaling, with elevated levels detected in estrogen receptor (ER)-positive breast tumors. 17 This estrogen-driven regulation highlights a potential link between HDAC6 expression and breast cancer metastasis. 18 Therefore, HDAC6 has emerged as a prognostic biomarker candidate, where increased expression may correlate with improved disease-free survival and responsiveness to endocrine therapy, emphasizing its importance as a therapeutic target. 18
The HDAC inhibitors have emerged as potential therapeutic agents for treating different cancers. These inhibitors are designed to induce cell cycle growth arrest, differentiation, apoptotic cell death, and alter protein expression. 19 As for now, 4 HDAC inhibitors are approved by the US Food and Drug Administration (FDA), which are vorinostat, belinostat, romidepsin, and panobinostat for the treatment of hematological cancers, while chidamide (tucidinostat) for different cancer treatment approved by China Food and Drug Administration. 20 A few classes of selective inhibitors are in clinical trials for various tumors. These drugs are classified as pan-HDAC inhibitors because they target multiple HDAC isoforms, which can lead to cell toxicity and side effects such as diarrhea, vomiting, and fatigue. 21 Till now, few selective HDAC6 inhibitors are in clinical trials such as ricolinostat (ACY-1215), citarinostat (ACY-241), and KA2507 for different tumors such as relapsed lymphoid malignancies, metastatic breast cancer, and melanoma. 22 These selective HDAC6 inhibitors are being used in combination therapies, having limited success as single target therapy. 22 Trichostatin A (TSN) is a natural pan-HDAC inhibitor targeting mostly class I and II HDAC isoforms for treating different cancer types. 23 The TSN is reported to reduce both the mRNA and protein expression levels of aromatase CYP19 phospholipase C gamma-1 in MCF-7 breast cancer cells.24,25 Many studies have reported that ricolinostat, as an HDAC6 inhibitor, is effective in reducing lymphoma cell growth and increasing apoptosis in combination with bendamustine; 26 promoting proteasome-mediated degradation in metastatic breast cancer patients in combination with nab-paclitaxel.27,28 Therefore, concerns have been raised about selective HDAC6 inhibitors as single agents with reduced adverse effects.
In recent years, significant progress in the discovery of selective HDAC6 inhibitors discovery has been made through novel scaffolds and advanced computational strategies. For instance, quinazolinylpropanoic acid derivatives targeting the ZnF-UBP domain have shown potent and selective HDAC6 inhibition with improved safety profiles. 29 The design and computational analysis of novel isatin-imine hybrids, notably Km68 and Km69 candidates, have shown strong HDAC6 binding and have favorable absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiles. 30 In parallel, artificial intelligence (AI)-assisted design pipelines have emerged, integrating structure-based docking and molecular dynamics (MD) refinement, combining deep learning to predict and validate structurally diverse HDAC6 selective compounds across large virtual hits. 31 Furthermore, machine learning models developed to predict selectivity between HDAC1 and HDAC6, compared 2 approaches: isoform-specific models and bioactivity difference models. The latter model showed superior performance in the identification of HDAC6-selective compounds across diverse chemotypes. 32
Despite the potential of HDAC6 as a therapeutic target, most clinically investigated HDAC6 inhibitors lack selectivity, leading to toxicity and off-target effects. 12 However, many computational studies rely on either small-scale screening libraries or omit long-timescale MD, 33 per-residue energy decomposition, or drug-likeness profiling. Few studies focus solely on docking-based screening without incorporating MD or per-residue energy decomposition, which are essential for protein flexibility and interaction stability overtime13,34 Moreover, drug-likeness and ADMET predictions are often underutilized or applied only post-screening in many computational studies, which limits their ability to prioritize compounds with favorable pharmacokinetic and toxicity profiles. 12 To further strengthen prospective predictivity, rigorous alchemical free energy methods, Free Energy Perturbation (FEP) and Thermodynamic Integration (TI) are used for top-ranked hits to obtain quantitatively robust affinity estimates and to cross-validate docking and molecular mechanics/generalized born surface area (MM/GBSA) results.
The study hypothesizes a computational multi-stage in silico pipeline for the identification of selective HDAC6 inhibitors in breast cancer. In silico drug discovery has become a cornerstone of modern pharmaceutical research, enabling the rapid screening of diverse chemical libraries, predicting target binding affinity, and evaluating drug-like prior to synthesis. These methods aid value in early-stage drug development, reduce experimental costs, and improve hit to lead identification.33,35 -38 In this study, the workflow integrates a large-scale virtual screening of 264 834 compounds with drug-likeness and reactivity filters, followed by induced-fit docking to optimize binding poses and account for protein-ligand flexibility. The top candidates were further evaluated through molecular dynamic simulations of 100 ns to access the stability of complex and conformational dynamics, and MM/GBSA binding free energy calculations with per-residue decomposition to identify key active site residues that have role in the protein-ligand interaction. Furthermore, comprehensive ADMET profiling was performed to predict pharmacokinetic properties, safety, and drug-likeness. This multistage strategy enables identification of candidates with improved selectivity, stability, and pharmacological suitability, thereby minimizing dependence on experimental resources during early phase screening.
Materials and Methods
Protein and ligand preparation
The X-ray crystallographic structure of human HDAC6 catalytic domain 2 in complex to TSN (PDB ID: 5EDU) with resolution 2.79 Å was selected from the RCSB protein data bank (https:www.rcsb.org/). 14 The HDAC6 protein was prepared using the protein preparation wizard of Schrödinger 2024-1 maestro. 39 The protein preparation steps are completed in 3 stages: The first is Pre-processing, where hydrogen atoms are added to the protein, filling of missing side chains and missing loops using prime refinement. The second is the Optimization of the H-bond using PROPKA at pH 7.0 for protein optimization, and the third step is Minimization, where the protein is minimized using the OPLS4 force field and deletion of water molecules that are beyond the active site. Ligands were selected from a library of chemical compounds built from different databases, including the Enamine (REAL) database, 40 ChemDiv, 41 InterBioscreen, 42 and the Halogen fragment library from Ottava 43 and Reaxense. 44 The compound libraries were pre-processed by removing duplicates. Duplicates were removed using RDKit, which is part of the Pre-LigPrep preparation process. During pre-processing, we enumerated physiologically relevant tautomers, ionization states, and stereoisomers for each unique input ligand. This approach enhanced chemical diversity while screening the library. In addition, we addressed analog redundancy and standardized structures as a post-processing step. The 3D compound libraries were prepared using LigPrep 45 with an OPLS4 force field that has enhanced accuracy in modeling protein-ligand interactions, conformational energetics, and solvation behavior. 46 Using Epik, ligand ionization states were generated at a pH range of 7.0 ± 2.0 (the general pH of biological systems) to generate multiple tautomers and protonation states for each ligand. Stereoisomer computation generates up to 32 possible stereochemical structures per ligand.
Virtual screening
Virtual screening is a computational technique to identify potential drug candidates from a large database of chemical compounds. The Fragment library and FDA-approved library collections from the Enamine database were selected, as this type of screening provides high-quality hits against proteins that can be classified as druggable compounds. ChemDiv consists of large-scale screening libraries and compounds for drug discovery. Many targeted libraries from ChemDiv, such as anticancer, antiviral, kinases, and epigenetic libraries, were chosen to find compounds that could bind to the protein and modify its activity. The InterBioscreen library consists of high-quality natural and synthetic compounds for high-throughput screening. The halogen fragment library comprises molecules with favorable halogen bond interactions in the binding sites. These libraries were screened using a virtual screening workflow in Schrödinger 2024-1. The threshold for selecting hits during virtual screening was based on Qikprop-predicted ADME properties, prefilter by the Lipinski rule via removal of ligands with the reactive functional group. These filters were applied to enrich for molecules with favorable oral bioavailability, membrane permeability, and overall drug-likeness, which are essential for anticancer therapeutic candidates. These ligands were finally docked with Glide XP (extra precision) 47 method. The final 1541 virtual hits from XP were selected based on glide gscore with a cutoff of −8.0 Kcal/mol, and the top twenty hits were validated using molecular docking studies.
Molecular docking
To better understand the specific interaction between HDAC6 protein and the top 20 hit-screened ligand molecules, docking was performed using Induced-fit Docking (IFD). The HDAC6 protein 5EDU was prepared using protein-preparation wizard. The docking experiment was validated with co-crystallized ligand TSN of the receptor to reproduce the crystallographic poses, and Ricolinostat, already a selective HDAC6 inhibitor, and the top 20 virtual hit compounds into the HDAC6 protein active site. The IFD application in Schrödinger 2024-1 uses rigid receptor docking (using Glide) and protein structure prediction and refinement (using Prime) methods to investigate the best conformational poses. 48 The HDAC6 protein was pre-processed and prepared using the default protein preparation wizard 39 with constrained minimization of the receptor with root mean square deviation (RMSD) to 0.30 Å. Initially, Glide docking of each ligand with receptor was performed by selecting the grid center dimensions as X: −1.24, Y: 8.71, Z: 5.83 Å and grid box size of 20 × 20 × 20 Å to encompass HDAC6 catalytic site using receptor grid generation. No specific constraints were applied, as the Zn+ ion present in the active site, coordinated by residues ASP649, HIS651, AND ASP742. Glide docking uses receptor and ligand van der Waals radii scaling as 0.50 and generates possible conformations of the ligand, and a maximum of 20 poses of each ligand were refined within 3.4 Å by Prime via rotamer-based library optimizations of the protein side-chain conformations. Glide redocking uses the XP method, as it combines a powerful sampling protocol with a custom scoring function designed to identify ligand poses that would be expected to have unfavorable energies 49 and redock into structure within 30 kcal/mol. Finally, the putative docked complexes and poses were ranked using the Glide score function and Prime energy. The top 4 best complexes were chosen based on the glide docking score and binding energies involved in the interaction of amino acids.
Molecular dynamics simulation
To explore the conformational changes in the protein and interaction of ligands within the binding complex, molecular dynamic simulation is performed using the Desmond module 50 in Schrödinger 2024-1. The simulation involves 3 main stages: (a) System Setup: A solvation model was constructed using the System Builder, where the SPC solvent model was applied with an orthorhombic boundary box shape size of 10 Å buffer on all sides. Sodium and chloride ions were added to neutralize the charges to 0.15 M NaCl concentration, and the OPLS4 force field was utilized. (b) Energy Minimization: Prior to MD simulation, the system underwent energy minimization to eliminate steric clashes and bring the structure to a local energy minimum, with minimization for up to 100 ps. (c) MD Simulation: System equilibration was conducted using the NPT ensemble at a temperature of 300 K and a pressure of 1.01325 bar. In addition, for temperature control Nosé-Hoover chain thermostat and for pressure Martyna-Tobias-Klein barostat were used. Following equilibration, a 100-ns simulation was executed for the protein-ligand docked complexes. The MD simulation studies for the top 4 hit docked complexes were conducted for a 100-ns run. The stability of protein-ligand complexes was analyzed by RMSD, root mean square fluctuations (RMSFs), and protein-ligand contacts by generating a simulation interaction diagram (SID) in the Desmond module. The final top 2 hits, HDI-2 and HDI-3, along with reference inhibitors, were run in replicates to account for stochastic variations and ensure reproducibility for more robust and reliable results.
Essential dynamics analysis
The essential dynamics of each protein-ligand complex trajectory were analyzed using Principal Component Analysis (PCA) and the Dynamic Cross-Correlation Matrix (DCCM). The PCA examines the conformational distribution throughout the MD simulation and analyzes the motions of proteins in the protein-ligand complexes. This analysis used the trj_essential_dynamics.py Python script from the Desmond program via the command line. The script performs essential dynamics analysis by calculating the protein’s C-alpha atoms’ principal components (PC) (eigenvectors). In PCA, the covariance matrix is constructed to capture the atomic positional deviations. The eigenvectors represent the PCs, indicating the direction of motion, while the eigenvalues correspond to the magnitude of these atomic fluctuations. This method helps elucidate dominant motions and conformational transitions in the protein-ligand complex. Correlated and anti-correlated motions were also analyzed, which are crucial for biological recognition and binding. These motions are derived from the covariance matrix of atomic fluctuations, calculated from the MD simulation trajectories. 51 The cross-correlation coefficient quantifies the degree of correlated movements between residue pairs. The script also computes the DCCM, generating a cross-correlation matrix visualizing the extent of correlated and anti-correlated motions. The DCCM results are plotted as frame-wise conformational deviations projected onto the calculated PC space. 52 The correlation coefficient ranges (r values) for residues i and j was calculated as:
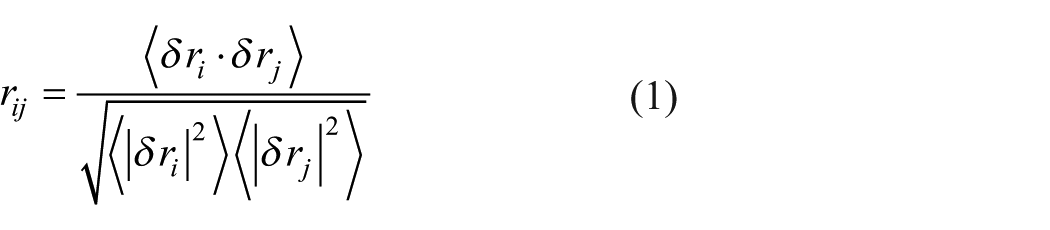
where
Binding free energy calculations
The free energy of the binding of ligands to biological macromolecules was calculated using MM/GBSA. The MM/GBSA is the fastest force-field-based method that computes the free energy of binding from the difference between the free energies of protein, ligand, and complex in solution. This method is based on the concept of a combination of molecular mechanics (MM) energies, polar and nonpolar solvation terms, and entropy terms that correctly render the approximate free energy of binding a ligand to a receptor. 54 For each complex, 20 frames were extracted from the final 100 ns of MD simulation trajectories. The MM/GBSA calculations were carried out for each frame using python script the thermal_mmgbsa.py and the average binding free energy along with standard deviation was computed to assess statistical reliability. Moreover, to measure the contribution of the individual residues, per-residue decomposition analysis provides the protein residue’s contribution in all possible binding modes throughout the simulation run. This was achieved using breakdown_MMGBSA_by_residue.py python script.
Druglikeness and in silico absorption, distribution, metabolism, excretion, and toxicity prediction
In silico methods for evaluating absorption, distribution, metabolism, and excretion (ADME) parameters rely on theoretical statistical models generated by correlating the structural characteristics of compounds tested in specific assays with their biological responses. These methods are now widely adopted due to their minimal resource requirements. The top 4 virtual hit compounds and known HDAC inhibitor TSN and Ricolinostat were assessed for their ADMET properties using the ADMETlab 2.0 55 web server (https://admetmesh.scbdd.com/).
Results and Discussion
Structure-based virtual screening to identify potential HDAC6 hits
Virtual screening is a computational technique to identify potential lead compounds by screening large libraries of chemical compounds. The potential hit compounds that bind to the HDAC6 protein target were identified using virtual screening. The different libraries screened are Fragment libraries and FDA-approved drug library from Enamine database; histone-targeted library, anticancer library, antiviral library, epigenetics and kinase library from ChemDiv database; natural compound library from InterBioscreen database; halogen fragment library from otava and reaxense databases. A total of 2 64 834 compounds were screened for the human crystal structure of HDAC6 (PDB: 5EDU). The reason for screening diverse libraries is that they represent a broad range of chemical structures, properties, and functionalities, and discover lead compounds that interact with specific biological targets. Potential hits were screened from different databases using a virtual screening workflow using Schrödinger 2024-1. The virtual screening was done using the XP method with glide, and filtering was done based on qikprop properties, the Lipinski rule of 5, and removing the ligands with reactive functional groups. The results were obtained with 1541 as the top virtual hit-screened compound based on glide gscore with a cutoff of −8.0 Kcal/mol and filtering parameters. Moreover, the involvement of important amino acids in the binding was noticed. The detailed results are provided in Supplemental Table 1, which lists the different chemical libraries screened, the initial number of compounds in each library, and the number of hits obtained after Glide XP virtual screening. When compared with previous HDAC6 virtual screening studies, our pipeline screened a substantially larger and more diverse data set, where single-library approaches12,17 were used to screen the compounds. Therefore, the top 20 virtual hit compounds, selected based on Glide gscores, favorable binding orientations, and predicted drug-like properties using Qikprop filters, were validated through induced-fit molecular docking against the HDAC6 protein to refine their binding poses and assess key protein-ligand interactions.
Molecular docking for binding affinity predictions
Induced-fit docking is a computational method to study the interactions between a ligand and a protein. The IFD accounts for the flexibility of both the ligand and the protein’s binding site. The IFD method determines the ligand’s preferred orientation within the protein’s binding site and estimates the binding affinity. The top 20 virtual hits were docked against the HDAC6 protein (PDB ID: 5EDU) using the IFD module in Schrödinger 2024-1. The docking experiment was validated by redocking the co-crystallized ligand TSN from the complex and Ricolinostat (a selective HDAC6 inhibitor) into the binding site of the HDAC6 protein. The RMSD of the reference ligand TSN and crystal structure of the protein was compared and found within the acceptable range (0.115 Å) as provided in Supplemental Figure 1. This was used for a comparative analysis with the results obtained from the virtual screening. In the Glide protocol, IFD typically involves a combination of molecular docking, protein structure refinement, and redocking steps to predict ligand binding modes accurately. Induced-fit docking studies aligned with the top 4 hit compounds from the virtual screening results. The docking scores for HDI-1 (−11.11 kcal/mol), HDI-2 (−11.73 kcal/mol), HDI-3 (−10.62 kcal/mol), HDI-4 (−11.57 kcal/mol), TSN (−10.83 kcal/mol), and Ricolinostat (−9.67 kcal/mol) as mentioned in Table 1. The table consists of glide gscore, which is a primary scoring function used in glide docking and estimates the binding affinity of the ligand to the protein; docking score evaluates the strength of interaction between ligand and protein during docking; glide energy indicates the stability of ligand-protein interaction; prime energy evaluates system energy post-refinement, accounting for protein flexibility and IFD score combines the docking and refinement energies to rank ligand-receptor complexes. 56 The HDI-2 showed better docking scores than the standards and the other 3 hit compounds. Also, a few similarities of amino acids are noticed in the binding patterns within the docked complexes. The HDI-3 has a low docking score compared to the standard TSN, but higher than the other standard Ricolinostat. Also, HDI-3 has high glide energy values, contributing to the binding affinity of the ligand to the protein. The ASP649, HIS651, and ASP742 form metal complexes with all 4 hit compounds and standard due to the presence of Zn+2 ions as a cofactor. The SER568 and TYR782 form hydrogen bonds with all selected hits except HDI-3 and HDI-4 dock complexes. The PHE680 residue is prominent in all docked complexes, forming hydrophobic interactions. The GLY619 residue forms hydrogen bonds with HDI-1, HDI-3, and HDI-4 complex, which is not observed in the standard TSN and HDI-2 complex. All the top 4 virtual hits have slightly better binding affinity when compared to both the standards, indicating a better stability in the HDAC6 protein binding site. The molecular docking study only provides insights into the interaction of the protein and ligand molecules. The glide docking scores offer a rapid and quantitative means of ranking ligands by its predicted binding affinity. These scoring functions estimate binding energy using ligand-protein interactions but do not fully capture conformational changes. 57 Therefore, further validation of these hit compounds can be explored using MD simulations and binding free energy calculations, which offers a more rigorous thermodynamic assessment. Figure 1 represents the 3D interaction profiles of the protein-ligand complexes, highlighting the key-active site residues engaged during docking and subsequent MD simulations.
Induced-fit docking results of HDAC6 with reference inhibitors (Trichostatin A and Ricolinostat) and top 4 screened hit compounds, summarizing binding scores and interaction energetics.


3D representation of protein-ligand interactions, showing (A) Trichostatin A, (B) Ricolinostat, (C) HDI-1, (D) HDI-2, (E) HDI-3, and (F) HDI-4.
Molecular dynamic simulation for stability assessment
For the MD simulation study, the docked best-scoring poses of these compounds were considered (selection was done depending on their docking scores and binding site interactions) with the protein HDAC6. All the trajectories of these docked complex structures were analyzed to understand the stability and fluctuations through RMSD, RMSF, and protein-ligand contacts for the simulation run of 100 ns (1-1000 frames). The RMSD and RMSF graphs were plotted using OriginPro 2023. 58 To enhance interpretability, improved interaction diagrams of molecular docking and post-MD complexes are represented as Supplemental Figure 2.
Root mean square deviation
The RMSD measures the structure’s average deviation from a reference conformation over the simulation run. It assesses the system’s stability and equilibration during the simulation run. The optimal range of RMSD is within ~1 to 3 Å. The RMSD of the protein C-alpha atoms provides insights into the structural conformations during the simulation run. The average protein RMSD values are observed as for TSN (3.6 Å), Ricolinostat (3.5 Å), HDI-1 (2.8 Å), HDI-2 (3.3 Å), HDI-3 (2.5 Å), and HDI-4 (2.5 Å) (Figure 2). The HDI-3 and HDI-4 are observed to have deviated at 200 to 400 frames (ie, 20-40 ns) and have stabilized toward the end of the simulation. Both the standards deviate most at 400 frames (40 ns), the same as with the HDI-2 hit, while the HDI-1 is found to deviate at 800 frames (80 ns). The ligand RMSD values show the ligand’s stability with respect to the protein. The average ligand RMSD values are for TSN (2.3 Å), Ricolinostat (9.4 Å), HDI-1 (3.1 Å), HDI-2 (2.7 Å), HDI-3 (2.8 Å), and HDI-4 (4.0 Å) (Figure 3). The ligand RMSD recommended range is ~1 to 4 Å relative to their initial binding pose in the protein. In such a case, Ricolinostat has attained a higher ligand RMSD than the top 4 virtual hits, indicating the ligand has an unstable binding in the binding pocket due to weaker interactions. All the complexes recorded a lower protein RMSD than the standard HDAC6 complexes. The protein RMSD deviations observed slightly above 3 Å for HDI-2, TSN, and Ricolinostat complexes are associated with flexible loops far from the binding pocket, and these do not disrupt the catalytic site geometry or ligand-residue interactions. The protein and ligand RMSD calculated for all 4 top hits were in the recommended range and favored a steady complex. However, HDI-3 is observed to have stabilized toward the end of the simulation, suggesting more steady stability than the other hit complexes.

Protein root mean square deviation (RMSD) of C-α atoms showing the structural deviations of HDAC6 protein in complex with (A) Trichostatin A (red), (B) Ricolinostat (yellow), (C) HDI-1 (blue), (D) HDI-2 (olive), (E) HDI-3 (purple), and (F) HDI-4 (green) over a number of frames (100 ns).

Ligand root mean square deviation (RMSD) analysis of HDAC6-ligand complexes. Ligand RMSD plots show positional deviations of (A) Trichostatin A (red), (B) Ricolinostat (yellow), (C) HDI-1 (blue), (D) HDI-2 (olive), (E) HDI-3 (purple), and (F) HDI-4 (green) within the HDAC6 binding pocket over 100 ns.
Root mean square fluctuations
The RMSFs specify the flexible regions of the protein and analyze the areas of structures that fluctuate most during the simulation run. The average protein RMSF values observed are for TSN (2.0 Å), Ricolinostat (1.2 Å), HDI-1 (1.5 Å), HDI-2 (1.6 Å), HDI-3 (1.4 Å), and HDI-4 (1.2 Å) (Figure 4). The N- and C-terminal regions of a protein typically exhibit more significant fluctuations compared to other parts. In contrast, secondary structure elements such as alpha helices and beta strands are generally more rigid, resulting in less fluctuation than the more flexible loop regions and unstructured parts of the protein. A high fluctuation was observed at residue THR807, with TSN, fluctuation at 6.4 Å and HDI-4 at 8.3 Å in Figure 4. Overall, low RMSF values were recorded for all complexes, but HDI-3 was observed to have fluctuated least and was stabilized toward the end of the simulation, suggesting greater stability than the other hit complexes.

Protein root mean square fluctuation (RMSF) analysis of HDAC6-ligand complexes. RMSF of C-α atoms is shown for HDAC6 residues in complex with (A) Trichostatin A (red), (B) Ricolinostat (yellow), (C) HDI-1 (blue), (D) HDI-2 (olive), (E) HDI-3 (purple), and (F) HDI-4 (green) over a 100-ns simulation.
Protein-ligand contacts
Protein-ligand contacts show the interaction between protein and ligand during simulation. These interactions are categorized into hydrogen bonds, hydrophobic, ionic, and water bridges. A similar interaction pattern was observed in all complexes, with the ionic interactions formed by ASP649, HIS651, and ASP742 residues throughout the simulation run. The HIS611 residue forms hydrogen bonds in HDI-2 and TSN complex, whereas for Ricolinostat, HDI-1, HDI-3, and HDI-4 complexes, GLY619 forms hydrogen bonding with more than 50% simulation time. Few additional residues are observed to make interactions, such as Asp567 makes multiple bonds like hydrogen, ionic and water bridges in HDI-1 complex, SER568 and TYR782 make hydrogen bonding with HDI-2 complex for more than 75% simulation time, PHE680 forms hydrogen and hydrophobic bonds with HDI-3 complex. The GLU779 residue forms strong hydrogen and slightly ionic bonds with the Ricolinostat and HDI-4 complexes. These residues mentioned above are necessary because of their interaction formed (Figure 5).

Protein-ligand contact analysis of HDAC6-ligand complexes. Protein-ligand contact profiles illustrate hydrogen bonds, hydrophobic contacts, ionic interactions, and water bridges formed between HDAC6 and (A) Trichostatin A, (B) Ricolinostat, (C) HDI-1, (D) HDI-2, (E) HDI-3, and (F) HDI-4 during 100 ns simulations.
The simulation experiments were further executed for 100 ns in triplicate for the top 2 hits, HDI-2 and HDI-3, as well as for reference inhibitors to ensure reproducibility for the results. Across replicates, HDI-3 maintained lower protein RMSD values consistently ~ 2.3 to 2.5 Å (Supplemental Figure 3) and a stable ligand RMSD profile as compared to HDI-2 and reference inhibitors, which displayed slightly higher deviations in certain runs (Supplemental Figure 4). Protein RMSF analysis (Supplemental Figure 5) showed HDI-3 has fewer fluctuations, induced less flexibility in key binding site residues, suggesting a more stable complex throughout the simulations. The HDI-3 consistently maintained stable ionic interactions with the key catalytic residues ASP649, HIS651, and ASP742 across all triplicate simulations for extended durations (Supplemental Figure 6).
Essential dynamics analysis of protein-ligand complexes
This method identifies and analyzes the dominant motions in MD simulations. It provides insights into protein structural dynamics and their functional implications. Essential dynamic analysis for the simulation trajectories for the top 4 hits was performed using PCA and dynamic cross-correlation matrices (DCCMs) in Schrödinger 2024-1. The PCA plot was generated using OriginPro 2023.
Principal component analysis
The PCA is a statistical technique used on simulation trajectories to identify and examine the primary motions within a biomolecular system. It showcases the dynamic behavior of the receptor molecule when ligands bind. For each MD simulation trajectory concerning the 4 most promising compounds, 10 PCs were produced using a Python script. The PC1, the primary component, captures the most significant large-scale conformational alterations, including protein domain movements and ligand binding/unbinding events. The second PC, PC2, represents secondary motions or correlated fluctuations in specific areas. The PC2 highlights localized conformational changes that are not apparent in PC1. Subtle movements, such as loop dynamics, solvent interactions, or side-chain rearrangements, are represented by higher-order PCs.34,51 A PCA plot was created using the top 2 components, PC1 and PC2, for both standard and the 4 leading compounds, focusing on protein C-alpha atoms (see Supplemental Figure 7). The PCA plots revealed that HDI-3 and HDI-4 formed tighter clustering in the PC1-PC2 space, suggesting more restricted conformational sampling of C-α atoms and strong residue correlations around Zn metal ion coordinating residues (ASP649, HIS651, ASP742). In contrast, HDI-2, HDI-1, TSN, and Ricolinostat show broader dispersion across PCs, suggesting higher flexibility and increased mobility leading to less stable binding interactions. The HDAC6 deacetylase activity required dynamic motions of the catalytic tunnel and surrounding loops to enable substrate entry and alignment. 14 Therefore, HDI-3 shows tight clustering and aligns with its lower RMSF and RMSD values, indicating the stabilized active site geometry contributing to improved inhibitory efficacy.
Dynamic cross-correlation matrices
The DCCM evaluates the correlated motions between residues over the simulation trajectory of 100 ns. The matrix plots the per-frame conformational deviation estimated on the calculated modes from the PC space. The heatmap diagrams are displayed as a color-coded matrix of Pearson correlation coefficients. The highly correlated motion is shown with positive values (blue region), while anti-correlated motions are shown with negative values (red region). The diagonal square relates to the correlation of residues with themselves. 52 The heatmap for DCCM explains the strong correlation between the motion of the standards and the top 4 hit compounds. The standard TSN shows highly correlated and anti-correlated motions. In contrast, Ricolinostat, HDI-1, HDI-2, HDI-3, and HDI-4 hits show changes across the motions. Highly correlated residue regions are 500 to 700 residues (yellow box), which shows the intense movement, suggesting the functionally active site residues. The DCCM heatmap shows mainly moderate correlations (r = 0.2-0.5) and weak anti-correlations (r = 2.0 to −0.4), indicating overall stability with limited large-scale motions. Stronger correlations (r > 0.6) were observed for HDI-3 and HDI-4 compared to standards and other hits. The DCCM heatmap of residue motions for standards and top 4 hits is provided in Supplemental Figure 8. These results can be further analyzed with binding free energy calculations and per-residue decomposition of active site residues for a more comprehensive conclusion, which will lead to the final hit selection.
Binding free energy calculations and per-residue energy decomposition using molecular mechanics/generalized born surface area
Binding free energy between a protein and a ligand is calculated using prime MM/GBSA. The MM/GBSA results show that all complexes’ binding energy (dG_bind) values are higher than those of both the standard complexes, except for HDI-4. The order of dG_bind values is exhibited as HDI-3 < HDI-1 < HDI-2 <Ricolinostat < Trichostatin A < HDI-4 (see Table 2). The HDI-3 compound has the highest negative binding free energy, −130.67 kcal/mol, while the HDI-4 compound possesses the least binding free energy, −110.83 kcal/mol. The HDI-1, HDI-3, and HDI-4 complexes are more involved in forming coulombic and hydrogen bond interactions than HDI-2 and the standards. All the HDI-1, HDI-3, and HDI-4 compounds have coulombic and hydrogen bond binding energy values higher than the standard, whereas Ricolinostat and HDI-2 have comparable values, suggesting them as stable complexes. These coulombic interactions and hydrogen bonds play critical roles in protein-ligand interactions and contribute to binding specificity, stability, and affinity. The detailed MM/GBSA binding energy results over the 100-ns simulations, calculated over 20 frames along with their corresponding dG binding energy averages values and standard deviations, are provided in Supplemental Table 2. The MM/GBSA is widely used for estimating binding free energies in protein-ligand systems, having several limitations. This method often omits configurational entropy contributions, resulting in overestimation of the magnitude of dG values. Furthermore, MM/GBSA employs an implicit solvent model, which cannot explicitly account for specific water-mediated interactions or long-timescale conformational changes. Therefore, to address these limitations, MM/GBSA calculations of the simulation trajectories were complemented with per-residue decomposition analysis, for a detailed and integrated assessment of protein-ligand interactions.54,59 The MM/GBSA calculations were performed for MD simulations in triplicate for the top hits HDI-2 and HDI-3 and reference inhibitors. Across replicates, HDI-2 exhibited favorable binding free energies, whereas HDI-3 was driven largely by strong coulombic interactions, comparable hydrogen bonds, and supportive van der Waals contributions with HDI-2 and reference inhibitors. The detailed replicate-wise average binding free energies are provided in the Supplemental Table 4.
MM/GBSA calculated binding free energies for HDAC6 in complex with reference inhibitors (Trichostatin A and Ricolinostat) and the top 4 hit compounds, describing the total dG_bind and individual energy components.

Abbreviations: dG_bind, binding free energy; dG_Coulomb, coulombic energy; dG_Cov, covalent energy; dG_Hbond, hydrogen bond energy; dG_Lipo, lipophilic energy; dG_solv, solvation energy; dG_vdw, van der Waals energy.
The binding free energies of individual residues contributing to ligand binding and active site amino acids were evaluated using per-residue decomposition. The important amino acid residues ASP649, HIS651, and ASP742 showed better coulombic interaction with binding free energies in all 4 hit compounds and the standard complexes. For TSN complex, the coulombic binding free energy for residues are ASP649 (−7.73 kcal/mol), HIS651 (−0.08 kcal/mol), and ASP741 (−7.1 kcal/mol); for Ricolinostat complex ASP649 (−8.1 kcal/mol), HIS651 (−0.34 kcal/mol), ASP742 (−8.15 kcal/mol); for HDI-1 complex possess ASP649 (−7.85 kcal/mol), HIS651 (−0.29 kcal/mol), ASP742 (−8.04 kcal/mol); for HDI-2 complex ASP (−7.79 kcal/mol), HIS651 (−0.27 kcal/mol), ASP742 (−8.06 kcal/mol); for HDI-3 complex ASP (−7.86 kcal/mol), HIS651 (−0.29 kcal/mol), ASP742 (−8.06 kcal/mol) and for HDI-4 complex ASP649 (−7.84 kcal/mol), HIS651 (−0.28 kcal/mol), and ASP742 (−7.96 kcal/mol). These 3 residues are critical, as they form ionic bond interactions in the binding of ligands to the protein. The HIS651 possesses comparatively low coulombic binding free energy, making multiple contacts with ligand compounds.
The Zn+2 ion involvement in the protein plays a significant role in binding as it forms solvation interactions with the compounds. The solvation binding free energies possessed by Zn+2 are for TSN −12.7 kcal/mol, Ricolinostat −13.07 kcal/mol, HDI-1 −12.8 kcal/mol, HDI-2 −12.6 kcal/mol, HDI-3 −12.9 kcal/mol, and HDI-4 −12.7 kcal/mol. The HIS610 residue is also involved in solvation interactions and has binding free energies as −0.24 kcal/mol for TSN, −9.86 kcal/mol for Ricolinostat, −9.92 kcal/mol for HDI-1, −9.82 kcal/mol for HDI-2, −10.08 kcal/mol for HDI-3, and −9.74 kcal/mol for HDI-4. The detailed pre-residue binding free energy decomposition results, including individual energetic contributions from key active site residues for all top 4 protein-ligand complexes, are provided in Supplemental Table 3, and the replicate-wise per-residue for the top 2 hits and reference inhibitors are provided in Supplemental Table 5 with key catalytic residues highlighted. The ligand molecule with the lowest binding energy is considered the most favorable, as it requires minimal energy to fit comfortably within the active pocket of the protein. Therefore, HDI-3 compound shows promising results in binding free energies and exhibits high coulombic interactions and hydrogen bond binding energy compared to the standards and other hit compounds.
Drug-likeness and in silico absorption, distribution, metabolism, excretion, and toxicity assessment for pharmacokinetic and safety profiling
To assess the properties of potential drug candidates, ADMET plays a crucial role in drug design. This computational approach helps filter out compounds with poor pharmacokinetics or high toxicity early in the drug discovery, saving time and resources. Based on the docking, simulation, and binding free energy results, the top 4 hit compounds were filtered for drug-likeness and pharmacokinetic properties using ADMETlab 2.0 web server. The ADMET evaluation from the ADMETlab web server predicts 17 physicochemical properties, 13 medicinal chemistry measures, 23 ADME endpoints, 27 toxicity endpoints, and 8 toxicophore rules. 55 A few critical ADMET properties of an anticancer drug molecule were identified and are discussed in Table 3. These properties are mentioned as Caco-2 permeability, MDCK (Madin-Darby Canine Kidney cells) cell permeability, Human Intestinal Absorption (HIA), Plasma Protein Barrier (PPB), Drug-Induced Liver Injury (DILI), AMES mutagenicity, hERG, Human hepatotoxicity, NR-ER (Nuclear Receptor-Estrogen Receptor), SR-p53, Nuclear Receptor-Peroxisome Proliferator Activated Receptor gamma (NR-PPAR-g), Carcinogenicity, Lipinski, Pan Assay Interference Compounds (PAINS), molecular weight (MW), and clearance (CL). The drug-likeness properties, such as LogP (a measure of hydrophobicity and lipophilicity), LogD (distribution coefficient), bioavailability score, topological polar surface area (TPSA), the Lipinski rule of 5, etc, for all 4 hit compounds predicted were in the range recommended as a drug candidate. The detailed ADMET results are provided in Supplemental Table 6, which contains the complete data set of predicted physicochemical, pharmacokinetic, and toxicity parameters of all hit compounds and reference inhibitors.
Predicted AMDET properties for the reference inhibitors (Trichostatin A and Ricolinostat) and the top 4 hit compounds, summarizing key pharmacokinetic, toxicity, and drug-likeness parameters.

Recommended range.
100-600.
Excellent (0-0.3), medium (0.3-0.7), and poor (0.7-1).
Excellent (⩾−5.15), poor (<5).
>2 × 10−6 cm/s.
⩽90%.
Excellent (⩾5) and poor (<5).
The in silico ADMET evaluation revealed that all selected hit compounds, along with the reference inhibitors TSN and Ricolinostat, demonstrated good predicted oral bioavailability. Among the 4 hits, HDI-3 displayed the most favorable profile with acceptable solubility (LogS = −2.336), good Caco-2 permeability and moderate HIA, all within the acceptable range for bioavailable anticancer drugs. Importantly, HDI-3 demonstrates low mutagenicity (Ames = 0.0012), reduced carcinogenicity risk (0.178) and no PAINS or chelating agents. For comparison, TSN and Ricolinostat were also predicted to have certain toxicological risks, including mutagenicity, Ames positivity, and respiratory toxicity alerts. These known inhibitors have experimental studies that support these predictions as Ricolinostat, reported in early clinical trials demonstrates good oral absorption and low hepatotoxicity. 60 While TSN is known to have genotoxic and mutagenic effects in vitro, 61 as reported experimentally, which align with our computational predictions. In contrast, HDI-2, despite its comparable structural stability in MD simulations, has several safety alerts, including PAINS, chelating agents, respiratory toxicity, Ames mutagenicity, and carcinogenicity. These findings suggest a higher likelihood of off-target binding and possible assay interferences, which could limit its progression as clinical lead. The ADMET analysis also predicts hERG inhibition (cardiotoxicity risk), hepatotoxicity, and nuclear receptor modulations, etc as extended beyond basic alerts. To validate these computational findings, in vitro toxicity studies including Ames mutagenicity, cytotoxicity assays, Caco-2 permeability assays, etc are recommended as a part of early phase experimental screening. Collectively, these findings indicate that HDI-3 combines a balanced pharmacokinetic profile with low toxicity risk, making it a stronger lead candidate for subsequent in vitro and in vivo validation.
Conclusion
The HDAC6 is a crucial metal-dependent enzyme playing a significant role in cellular regulation and is an essential target for epigenetic cancer therapy. In this study, we adopted a comprehensive computational strategy that included virtual screening, docking, MD, and MM/GBSA calculations to identify selective HDAC6 inhibitors for breast cancer treatment. Out of 4 promising candidates, HDI-3 exhibited the highest binding affinity, stable interactions with key residues, and consistent structural stability throughout 100 ns simulations. In-depth dynamics analysis further established that HDI-3 provides enhanced protein-ligand stability compared to standard inhibitors. The MM/GBSA binding free energy and in silico ADMET profiling highlighted HDI-3’s encouraging drug-like characteristics, such as excellent bioavailability and minimal toxicity risks. Although the computational findings are promising, experimental validation is crucial to address biological complexities, entropic effects, and long-term safety considerations. Future in vitro and in vivo studies are essential to confirm the efficacy, selectivity, and safety of HDI-3 as a targeted HDAC6 inhibitor for breast cancer therapy. This research underscores the importance of integrated computational methods in fast-tracking early drug discovery.
Future perspectives
The findings of the study provide a strong foundation for the HDAC6 inhibitor identification, where HDI-3 emerges as the promising lead due to its structural stability, favorable binding energies, and clean ADMET profile. Replicate MD and MM/GBSA studies showed comparable stability and binding trends for both HDI-2 and HDI-3 compounds. However, HDI-3 holds a slight advantage due to stronger ionic and hydrogen bonds with key catalytic residues and safer ADMET profile. Both HDI-2 and HDI-3 will be considered for wet-lab validation through in vitro enzymatic assays, cell-based studies, cytotoxicity assays, and in vivo evaluations. Future computational work will be focused on extended simulations and improved free energy methods such as TI and FEP to refine relative binding affinity predictions.
Limitations
The current study presents a comprehensive multi-stage in silico workflow for the identification of HDAC6 inhibitors, certain limitations are to be acknowledged. The results are based entirely on computational predictions and experimental validation (in vitro enzymatic assays, cell-based studies, and in vivo toxicity tests) is essential to confirm the findings. Virtual screening validation methods, like enrichment factor and receiver operating characteristic (ROC)-based metrics incorporation, could strengthen the robustness of the screening protocol. The MD simulations and MM/GBSA experiments were run for 100 ns in replicates to increase robustness, both showing consistent trends for the hits. However, running the simulations for longer or advanced sampling approaches could provide more exhaustive view of conformational landscape. In addition, ADMET predictions were based on statistical models and could not fully capture complex in vivo pharmacokinetics and toxicity profiles; hence, they should be experimentally verified.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-docx-1-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-2-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-3-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-3-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-4-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-4-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-5-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-5-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-6-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-6-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Supplemental Material
sj-xlsx-7-bbi-10.1177_11779322251379037 – Supplemental material for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling
Supplemental material, sj-xlsx-7-bbi-10.1177_11779322251379037 for Identification of Potent HDAC6 Inhibitors for Breast Cancer Through Multi-Stage In Silico Modeling by Vaishali Pankaj, Inderjeet Bhogal and Sudeep Roy in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
This research was supported by the Department of Biomedical Engineering, Faculty of Electrical Engineering and Communication, Brno University of Technology, Brno, Czech Republic.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability
The data sets generated and analyzed are provided in the manuscript supporting this study and are available in the supplementary materials.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.