
Abstract
Hepatocellular carcinoma (HCC) remains a major cause of cancer-related mortality worldwide. Aurora kinase A (AURKA), a critical regulator of mitosis and spindle assembly, has been implicated in tumorigenesis, yet its clinical relevance and immune associations in HCC require further clarification. Here, we performed integrated bioinformatic analyses using TCGA/GTEx-derived datasets and public platforms (including TIMER and GEPIA2) to characterize AURKA expression patterns, prognostic significance, and functional pathways in HCC. Potential upstream noncoding RNA (ncRNA) interactions were explored using ENCORI and miRNet 2.0, and immune microenvironment correlations were assessed via TIMER/GEPIA2 and TISIDB. We found that AURKA was significantly overexpressed in HCC and associated with unfavorable prognosis and more advanced clinicopathological features. A putative ncRNA regulatory axis involving DUXAP8 and hsa-miR-490-3p was identified as being associated with AURKA expression. Moreover, AURKA expression correlated with immune cell infiltration and immune-related features, suggesting a relationship with the tumor immune microenvironment. Collectively, our analyses suggest that a putative DUXAP8/hsa-miR-490-3p/AURKA regulatory network may be associated with HCC progression and correlates with features of the tumor immune microenvironment. These findings offer insight into potential biomarkers and therapeutic targets for personalized HCC treatment.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is a highly aggressive primary malignancy of the liver, frequently presenting with an insidious onset and poor clinical outcomes. 1 Despite substantial advances in diagnostic techniques and therapeutic strategies, the 5-year survival rate of HCC remains disappointingly low, largely attributable to its high propensity for distant metastasis.2,3 The major etiological factors driving hepatocarcinogenesis include chronic hepatitis B and C virus infections, aflatoxin exposure, and nonalcoholic steatohepatitis.4,5 Current treatment modalities for HCC comprise liver transplantation, surgical resection, transcatheter arterial chemoembolization, targeted therapy, yttrium-90 radioembolization, radiofrequency ablation, immunotherapy, and systemic chemotherapy. 6 However, these interventions have yielded only limited improvements in long-term survival, underscoring an urgent need to elucidate the molecular mechanisms underlying HCC progression and to identify novel therapeutic targets.
Aurora kinases are a family of serine/threonine kinases consisting of Aurora A, B, and C, which play essential roles in regulating mitotic events and maintaining genomic stability.7,8 Among them, Aurora kinase A (AURKA) has been extensively implicated in tumorigenesis, with aberrant expression and genetic alterations reported across a wide spectrum of human malignancies.9,10 Functionally, AURKA participates in both DNA synthesis and mitotic progression, thereby contributing to uncontrolled cell proliferation and cancer development.9,11
Also known by several aliases—including AIK, AURA, ARK1, BTAK, STK7, STK6, STK15, and PPP1R47—AURKA plays a central role in cell cycle regulation by orchestrating microtubule nucleation, centrosome maturation and separation, bipolar spindle assembly, and spindle pole stabilization during chromosome segregation.12,13 Although AURKA is expressed at relatively low levels in most normal tissues, with notable expression in the testis and lymph nodes, it is frequently overexpressed in a variety of tumors, particularly in highly proliferative cancer cells.14,15 Accumulating evidence indicates that AURKA is markedly upregulated in HCC and is closely associated with unfavorable clinicopathological characteristics and poor prognosis.12,16 Moreover, experimental studies have demonstrated that AURKA knockdown enhances radiosensitivity in colorectal cancer cells, highlighting its potential as a therapeutic target. 17 Nevertheless, the precise biological functions and clinical significance of AURKA in HCC remain incompletely defined.
Accordingly, this study aimed to clarify the clinical and immunological significance of AURKA in HCC. We comprehensively analyzed AURKA mRNA and protein expression, its associations with clinicopathological features, and its prognostic value, while further exploring AURKA-related biological functions, signaling pathways, and a putative ceRNA regulatory network. In addition, we assessed the relationship between AURKA expression and the tumor immune microenvironment (TIME), including immune cell infiltration and immune checkpoint–related characteristics, with the goal of identifying an AURKA-centered regulatory framework with potential implications for biomarker development and immunotherapy-oriented stratification in HCC.
Materials and Methods
Omics analysis of AURKA
This study aimed to elucidate the oncogenic role of AURKA in HCC and to explore potential carcinogenic mechanisms. Gene-related information, including gene nomenclature, identifiers, and chromosomal localization, was obtained from the NCBI Gene and Protein databases (Human Protein Atlas, https://www.proteinatlas.org/). The UniProt database was used to analyze the secondary structure and conserved domains of AURKA, which were visualized using the Illustrator for Biological Sequences tool. Subcellular localization of AURKA was examined based on immunofluorescence data from the Human Protein Atlas. Sequence conservation and evolutionary relationships were assessed using NCBI HomoloGene and COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/) to construct a phylogenetic tree.
Expression analysis of AURKA in pan-cancers
We first examined AURKA mRNA expression in normal human tissues using the HPA database. Then, using TIMER (http://timer.comp-genomics.org/), we compared AURKA expression between cancer and adjacent normal tissues across various cancer types. GEPIA2 (https://gepia2.cancer-pku.cn/#analysis) was also used to analyze AURKA expression across different cancers and normal tissue regions, based on data from TCGA and GTEx. A log2-fold change ⩾1 and P value <.01 were considered as criteria for differential expression.
Prognostic analysis of AURKA in pan-cancers
Kaplan-Meier survival analyses were performed to assess the prognostic significance of AURKA expression across 13 cancer types using data from the GEPIA2 database, with disease-free survival (DFS) and overall survival (OS) as endpoints. Statistical significance was defined as P < .05. In addition, the association between AURKA expression and clinical outcomes was further evaluated across multiple clinicopathological subgroups using the Kaplan–Meier Plotter database, including tumor stage and grade, vascular invasion, American Joint Committee on Cancer T classification (AJCC T classification), patient race, hepatitis virus status, alcohol consumption, and sex. Results were summarized and visualized using forest plots.
AURKA expression and clinicopathological features
The GEPIA2 Liver Hepatocellular Carcinoma (LIHC) dataset was used to assess the association between AURKA expression and clinicopathological stage in HCC. In parallel, the UALCAN (http://ualcan.path.uab.edu/) database was employed to analyze correlations between AURKA mRNA expression and clinicopathological features, including metastasis, tumor stage, and grade. Statistical significance was defined as P < .05.
Candidate upstream miRNA of AURKA prediction
The ENCORI database (https://starbase.sysu.edu.cn/) was used to predict upstream miRNAs targeting AURKA by integrating 7 algorithms, and miRNAs identified by at least 3 tools were selected. miRNA-mRNA interaction networks were constructed using Cytoscape, and correlations between AURKA and candidate miRNAs were assessed using Spearman analysis. Three miRNAs showing significant negative correlations with AURKA expression were identified, and their expression patterns and prognostic relevance in LIHC were further evaluated using the TCGA module of the UALCAN database.
Candidate upstream lncRNAs of AURKA prediction
The miRNet 2.0 database (https://www.mirnet.ca/miRNet/home.xhtml) was used to predict upstream long noncoding RNAs (lncRNAs) targeting hsa-miR-490-3p, and lncRNA-miRNA interaction networks were visualized using Cytoscape. Differential expression and prognostic analyses were performed using the GEPIA database, and lncRNAs positively associated with HCC were selected for further analysis. Associations between candidate lncRNAs and clinicopathological stage were evaluated across HCC stages, and ENCORI was subsequently used to assess the co-expression of hsa-miR-490-3p with AURKA and DUXAP8.
Immune cells infiltration, immune cell biomarkers, and immune cell related chemotactic activities analysis of AURKA
In this study, the TIMER tool was used to assess the correlation between AURKA expression and immune cell infiltration levels. We then focused specifically on HCC samples, where AURKA serves as a significant, independent unfavorable prognostic biomarker and is strongly linked to immune infiltration. In addition, both the TIMER and GEPIA2 databases were used to evaluate the relationship between AURKA expression and markers related to chemotaxis and immune cells.
Immune checkpoints genes analysis of AURKA
TISIDB (http://cis.hku.hk/TISIDB/) is an online platform that helps to study of interactions between the immune system and tumors. In our study, we used the TISIDB database to investigate the relationship between AURKA expression and immunoinhibitors. Furthermore, we explored the associations between AURKA expression and immune subtypes in HCC tissues.
Functional enrichment analysis
We used GeneMANIA (http://genemania.org/) to construct protein-protein interaction networks and predict the biological function of co-expressed genes. Subsequently, we obtained AURKA-correlated genes from the LinkedOmics (http://www.linkedomics.org/) database. We then conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses on these AURKA-correlated genes also using LinkedOmics as well.
Expression validation based on external datasets
We obtained gene expression profiles from the HCCDB (http://lifeome.net/database/hccdb/home.html) database and added them to our analysis. To visualize the differential expression level of AURKA between HCC tissue and normal tissues, we generated a box plot. In addition, protein expression levels of AURKA in HCC and normal tissues were determined using immunohistochemical staining based on the HPA database.
Results
Omics analysis of AURKA
The main objective of this study was to investigate the carcinogenic effects of human AURKA gene on HCC. The AURKA gene (Gene ID: 6790) is located on chromosome 20q13.2 and consists of 12 exons (Figure 1A). To better understand its biological function, the predicted secondary structure of the AURKA protein is demonstrated in Figure 1B. The AURKA gene gives rise to 2 major protein isoforms, AURKA isoforms X1 and X2, which are predominantly localized to key mitotic and cytoplasmic compartments, including the nucleoplasm, centrosomes, mitotic spindle apparatus, and cytosol in human cells (Figure 1C). These isoforms have been observed in human skin carcinoma cell (A-431) and human osteosarcoma cells (U-2 OS) (Figure 1D and E). Our study confirms that AURKA is a highly conserved member of the kinase family that contains one conserved domain, the serine/threonine protein kinase catalytic domain (S_TKc, smart00220), located on 127-384 (Figure 1A). The phylogenetic tree of AURKA protein presented the evolutionary relationship of these Opisthokonta species, and it was produced using the fast minimum evolution method (Figure 1F). These results indicate that AURKA protein likely plays a crucial role in different species.
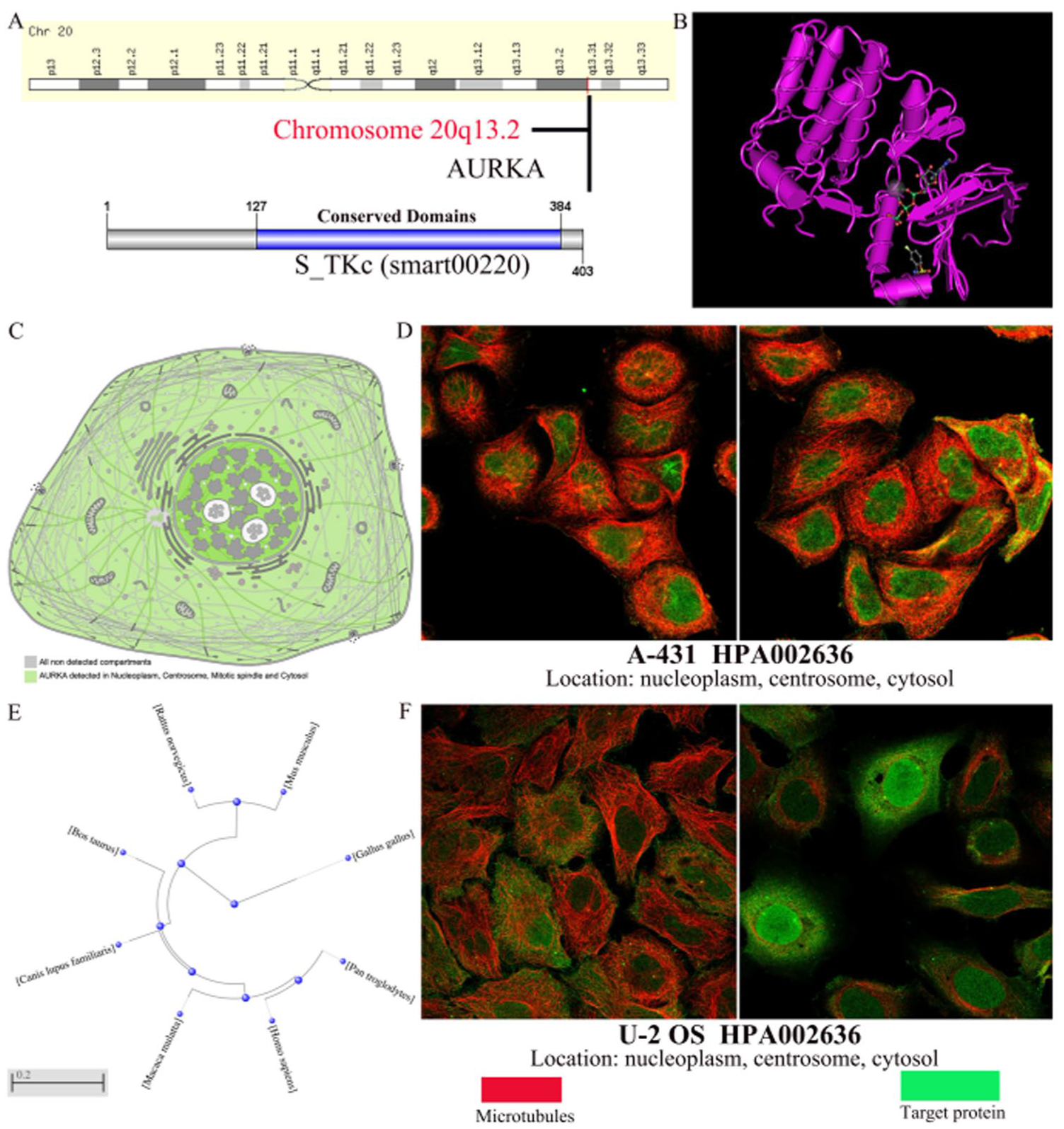
Genomics information of the AURKA. (A) Positional information of AURKA genes on chromosomes and the locations of the conserved domains of AURKA genes. (B) The secondary structure of AURKA protein. (C) Localization of AURKA protein in human cells. (D, F) Localization of AURKA in (D) human skin carcinoma cell (A-431) and (F) human osteosarcoma cells (U-2 OS) were assessed by immunofluorescent staining. (E) Phylogenetic trees of AURKA homologues.
Analysis of AURKA expression across pan-cancer
To characterize the baseline expression pattern of AURKA, we first analyzed its transcriptional levels across 27 normal human tissues using data from the GTEx database. The analysis demonstrated that AURKA expression was most pronounced in testicular tissue, reaching 21.5 normalized transcripts per million (nTPM), markedly exceeding levels observed in other tissues (Figure 2A). In addition, AURKA expression in normal liver tissue reached 5.0 nTPM, representing a relatively higher baseline level compared with most other normal tissues (Figure 2A). To further investigate the potential oncogenic involvement of AURKA in tumor development, we subsequently analyzed its expression profiles across multiple cancer types using the TIMER database (Figure 2B). We integrated GTEx normal tissue data with TCGA tumor datasets to validate AURKA expression. (Figure 2C). In summary, our findings suggest that the expression of AURKA in tumors, including colon adenocarcinoma, head and neck squamous cell carcinoma, HCC, lung squamous cell carcinoma, stomach adenocarcinoma, uterine corpus endometrial carcinoma, cholangiocarcinoma, esophageal carcinoma, rectum adenocarcinoma, breast invasive carcinom, bladder urothelial carcinoma, lung adenocarcinoma, and kidney chromophobe, was higher than that in corresponding paracancerous tissues.
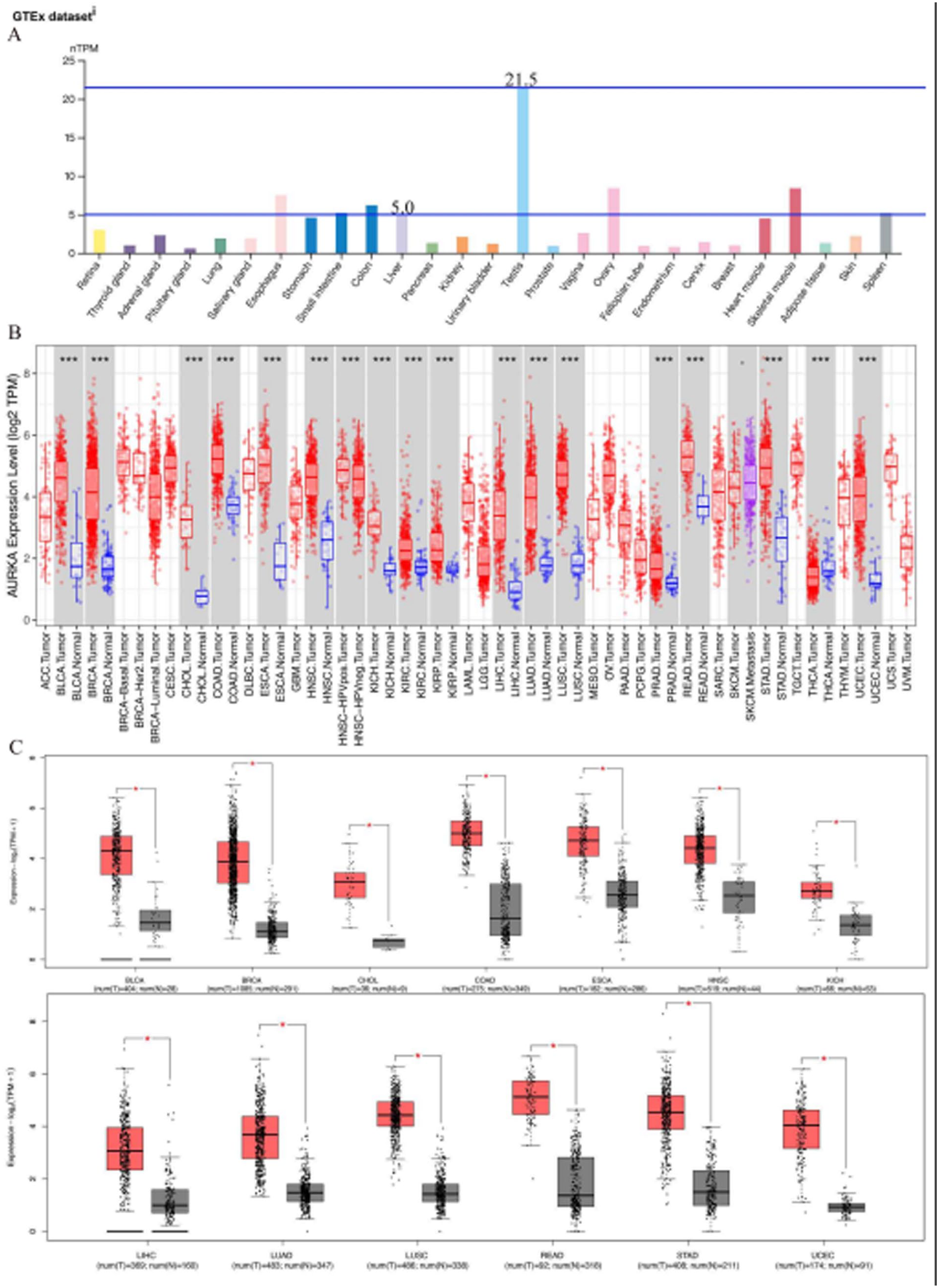
Expression of AURKA in normal and tumor tissues. (A) Expression of AURKA in normal human tissues. (B) The differential expression levels of AURKA between tumor and normal tissues in various tumor types in TIMER database. (C) The differential expression levels of AURKA between tumor and normal tissues in various tumor types in GEPIA2 database.
The prognostic value of AURKA in human cancers
GEPIA2 was used to assess the relationship between AURKA expression and clinical prognostic value in 13 types of cancer (Supplemental Figure S1A and B). Survival analyses demonstrated that AURKA expression was significantly correlated with both OS and DFS in patients with HCC. Specifically, elevated AURKA expression was associated with significantly poorer OS and DFS compared with low expression levels. To further delineate its prognostic relevance, subgroup analyses were performed to evaluate the relationship between AURKA expression and survival outcomes across distinct clinicopathological strata in HCC. The results were consistent across most of the clinicopathological characteristics subgroups, indicating that high AURKA expression was strongly correlated with poor prognosis, whereas low AURKA expression was associated with better prognosis (Supplemental Figure S1C and D).
Prediction of miRNAs upstream of AURKA
There is increasing evidences supporting that noncoding RNAs (ncRNAs) play significant roles in cancer progression by regulating oncogene expression. 18 To investigate the potential regulatory relationship between AURKA and miRNAs, we used the ENCORI online database to predict the miRNAs upstream of AURKA and identified 28 candidate miRNAs. The AURKA-miRNA regulatory relationship pair network was established and visualized with Cytoscape software (Figure 3A). Based on the theory of competing endogenous RNA (ceRNA) regulatory network, 19 we hypothesized that hsa-let-7c-5p, hsa-let-7g-5p, and hsa-miR-490-3p may be the upstream miRNAs of AURKA and regulate its expression(Figure 3B). We further explored the expression status and prognostic value of these upstream miRNAs based on the UALCAN database. Our results showed the expression level of hsa-let-7c-5p, hsa-let-7g-5p, and hsa-miR-490-3p was markedly downregulated in HCC (Figure 3C to E). Only the upregulated hsa-miR-490-3p was associated with a positive prognosis for patients with HCC (Figure 3F to H). Therefore, we concluded that hsa-miR-490-3p may be an upstream regulator of AURKA expression in HCC.
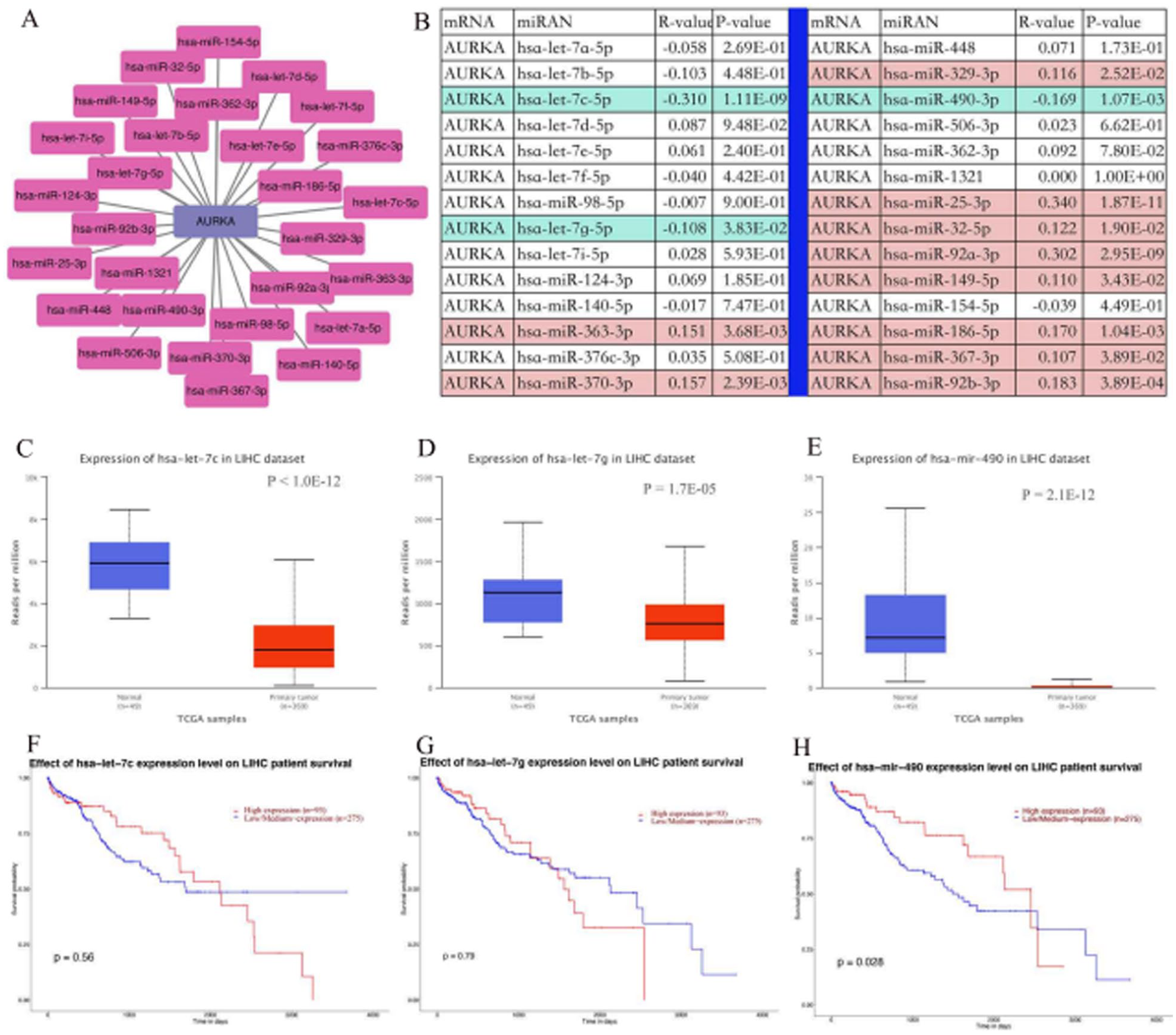
Identification of hsa-miR-490-3p as a potential upstream miRNA regulating AURKA expression in HCC. (A) Construction of miRNAs-AURKA regulatory network using Cytoscape software. (B) Spearman correlation analysis was employed to evaluate the correlation between predicted miRNAs expression and AURKA expression. (C, D, and E) Expression level of hsa-let-7c-5p (C), hsa-let-7g-5p (D), and hsa-miR-490-3p (E) in HCC and normal tissues was analyzed with TCGA dataset by using UALCAN database. (F, G, and H) Prognostic analysis of hsa-let-7c-5p (F), hsa-let-7g-5p (G), and hsa-miR-490-3p (H) in patients with HCC was done with TCGA dataset by using UALCAN database.
Prediction and analysis of lncRNAs upstream of hsa-miR-490-3p
The miRNet2.0 online tool was used to predict the upstream lncRNAs of hsa-miR-490-3p, resulting in the identification of 44 potential upstream lncRNAs. The lncRNAs-hsa-miR-490-3p regulatory relationship pair network was visualized using Cytoscape software (Figure 4A). Among them, only DUXAP8 was found to be highly expressed in tumor tissues compared with the adjacent normal controls (Figure 4B), and its upregulation was negatively correlated with prognosis (Figure 4C). Moreover, the expression level of AURKA is found to be associated with clinicopathological stages (Figure 4D). Subsequently, we investigated the correlation between the AURKA and hsa-miR-490-3p as well as DUXAP8 using the ENCORI database (Supplemental Figure S2A to D). According to the ceRNA theory, DUXAP8 could be the most promising upstream lncRNA for the hsa-miR-490-3p /AURKA axis in HCC.

Identification of DUXAP8 as a potential upstream lncRNA regulating hsa-miR-490-3p expression in HCC. (A) Construction of lncRNAs-hsa-miR-490-3p regulatory network using Cytoscape software. (B) Differential expression analysis of DUXAP8 in normal tissues and HCC tissues. (C) Prognostic analysis of DUXAP8 in patients with HCC. (D) The correlation between DUXAP8 expression and clinicopathologic stages.
Immune cells infiltration and chemotactic activities analysis of AURKA in HCC
To study the effects of differential AURKA expression on different tumor-infiltrating immune cells (TIICs), we explored the relationship between AURKA expression and different TIICs in HCC. As shown in Supplemental Figure S3A to F, AURKA expression showed a significantly positive correlations with the infiltration levels of B cells (partial.cor = .424, P = 2.01E-16), CD8+ T cells (partial.cor = .258, P = 1.31E-06), CD4+ T cells (partial.cor = .135, P = 1.24E-02), macrophage (partial.cor = .301, P = 1.42E-08), neutrophil (partial.cor = .308, P = 4.99E-09), and dendritic cells (partial.cor = .340, P = 1.18e-10) in HCC.
Expression correlation of AURKA and biomarkers of immune cells in HCC
To further validate the association of AURKA expression with tumor-related immune cell infiltration in HCC, we conducted a correlation analysis between the AURKA expression and well-known immune-related biomarkers of immune cells, including CD8+ T cells, B cells, neutrophils, CD4+ T cell, M2 macrophages, M1 macrophages, and dendritic cell (Table 1). Using the GEPIA2 database, we found that AURKA expression was significantly correlated with the most immune-related biomarkers of tumor-related immune cells, including neutrophil, CD8+ T cell, M2 macrophage, CD4+ T cell, dendritic cell, and M1 macrophage. These results were similar in the TIMER database as well. Overall, these findings demonstrate that AURKA is critical for tumor-related immune cells infiltration in HCC.
Correlation analysis between AURKA and biomarkers of immune cells in HCC determined by GEPIA and TIMER databases.
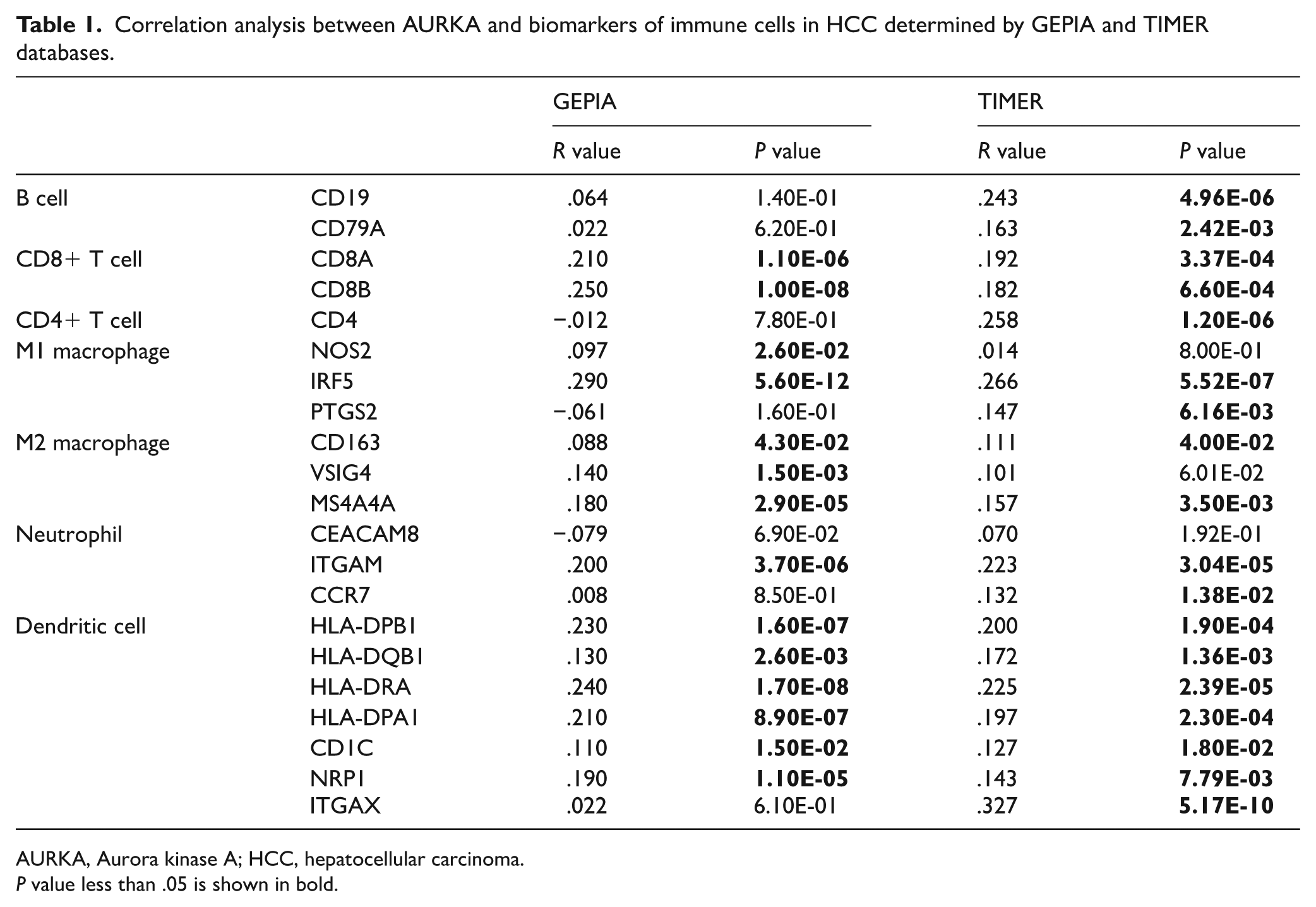
AURKA, Aurora kinase A; HCC, hepatocellular carcinoma.
P value less than .05 is shown in bold.
Correlation between AURKA expression and immune checkpoints in HCC
Cancer immunotherapy is increasingly being used to activate the immune system to treat tumors, and PD-1/PDL1 and CTLA-4 being the most appropriate targets for cancer immunotherapy. 20 To assess the effect of AURKA expression on HCC immunotherapy, we evaluated the relationship between AURKA expression and immune checkpoints using the TISIDB platform. The results showed that AURKA expression status correlated with CD96 (r = −.115, P = 2.69E-02), CSF1R (r = −.159, P = 2.16E-03), CTLA4 (r = .159, P = 2.06E-03), KDR (r = −.336, P = 3.34E-11), LAG3 (r = .133, P = 1.04E-02), PDCD1LG2 (r = −.118, P = 2.31E-02), TGFB1 (r = −.111, P = 3.22E-02) (Figure 5A to H). We also investigated the role of AURKA in immune subtypes in HCC using the TISIDB website. As expected, AURKA expression was significantly different in various immune subtypes (P = 1.77E-09) (Figure 5I). Thus, these results imply that AURKA expression can, as a critical soluble factor, influence the antitumor immune response.
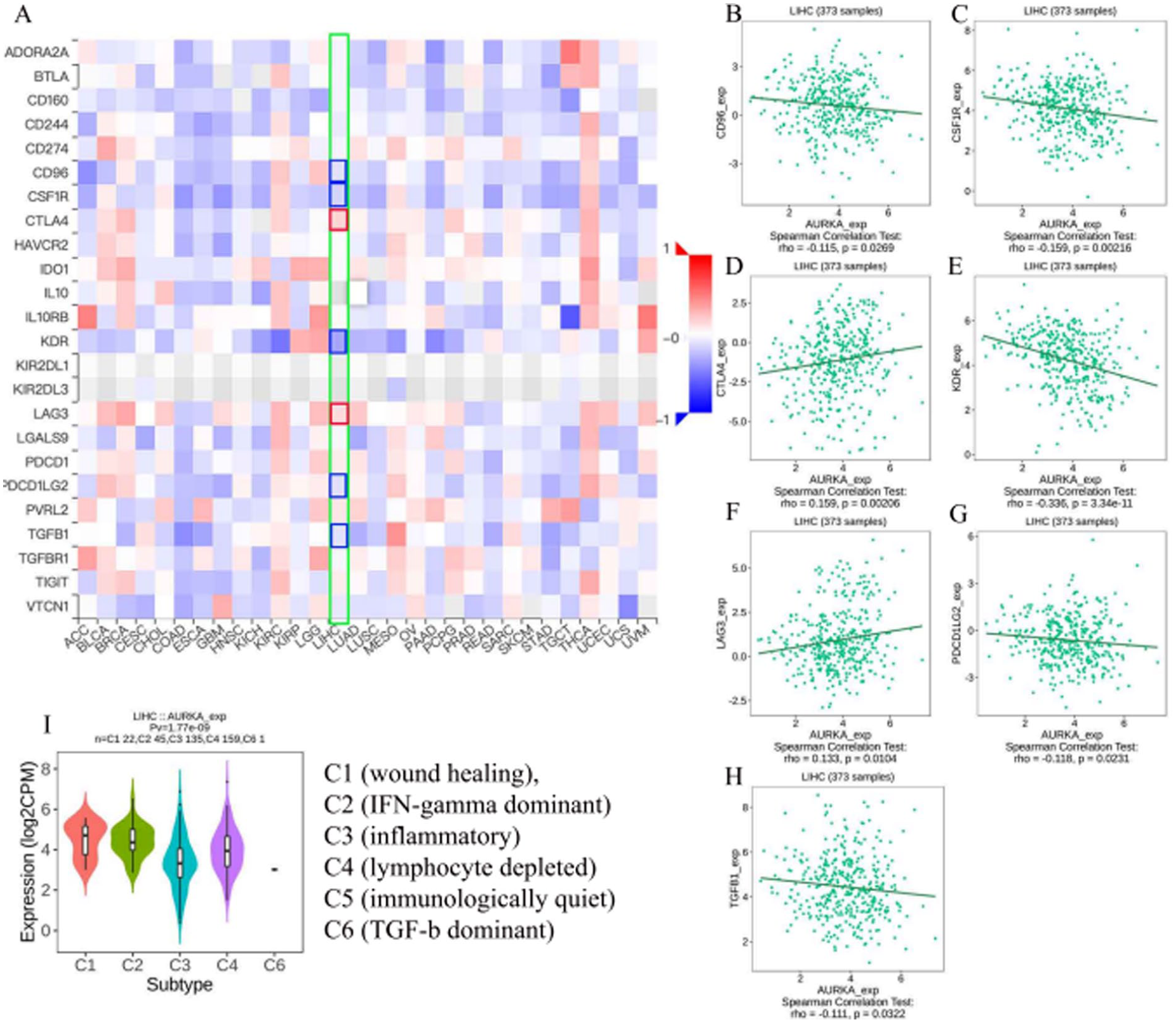
Correlation between AURKA expression level and immunomodulators in HCC from TISIDB database. (A) The hotmap presented the correlations between the AURKA and immunomodulators in pan-cancer. (B-H) The scatter plot shows the significant correlations between AURKA expression and CD96 (B), CSF1R (C), CTLA4 (D), KDR (E), LAG3 (F), PDCD1LG2 (G), TGFB1 (H). (I) The relationship between immune types and AURKA expression.
Functional analysis of AURKA
To investigate the underlying biological mechanism of AURKA in tumor development, we explored a network of AURKA and related gene sets using GeneMANIA. The results showed that 20 genes, including ZNF239, CHFR, CPEB1, DLGAP5, CDC37, AURKAIP1, TACC3, TPX2, HMMR, PLK1, CEP192, JTB, INCENP, AJUBA, FBXL7, BORA, WRN, TCF3, SOX3, and TDRD7, were primarily related to the function of AURKA. Most of these genes play roles predominantly in metabolism of spindle organization, microtubule cytoskeleton organization involved in mitosis, cell cycle G2/M phase transition, mitotic nuclear division, and spindle (Supplemental Figure S4A). Next, we examined the co-expressed genes of AURKA in the HCC cohort by LinkedOmics. As shown in Supplemental Figure S4B, green dots represent genes were negatively correlated with AURKA expression, whereas red dots denote genes that were positively correlated with AURKA expression. The heatmaps presented the top 50 significant gene sets positively and negatively linked with AURKA expression (Supplemental Figure S4C and D). The GO analysis demonstrated that the co-expressed genes of AURKA mainly participated in DNA replication, chromosome segregation, and so on (Supplemental Figure S4E). The KEGG pathway analysis indicated that the co-expressed genes of AURKA were mainly enriched in the oocyte meiosis, cell cycle, and DNA replication, and so on (Supplemental Figure S4F). Taken together, these results suggest that upregulation of AURKA in HCC may significantly contribute to tumor development by affecting key cellular processes involved in cell division and chromosome organization.
Expression verification
To comfirm the above findings, 8 datasets were analyzed, including 7 GEO datasets and 1 ICGC dataset, which demonstrated a significantly increase in AURKA mRNA expression in HCC compared with paracancerous tissue (Figure 6A). In addition, immunohistochemistry studies were conducted, which confirmed that high AURKA protein expression was higher in HCC tumor tissues than normal liver tissues (Figure 6B and C).
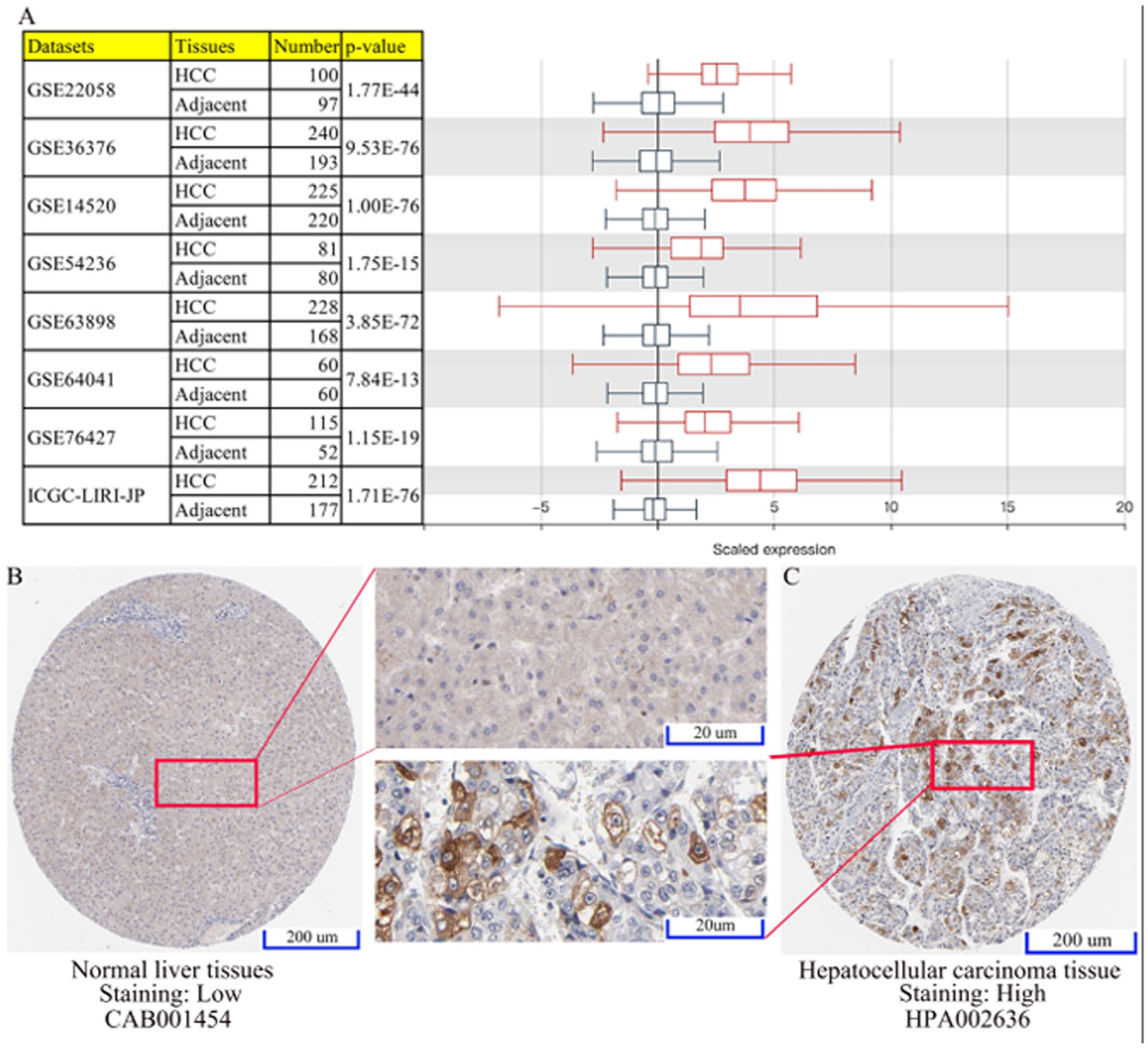
The AURKA expression was evaluated in HCC tissue/cells and normal tissue/cells based on the results obtained from different datasets. (A) Boxplot for the AURKA expression profile across cancer type generated with the GENT2 database. (B-C) The expression of AURKA protein in HCC and normal sample was detected by immunohistochemistry from the HPA database.
Discussion
Hepatocellular carcinoma ranks among the leading causes of cancer-related mortality worldwide, accounting for substantial global cancer deaths. 2 Increasing evidence indicates that the TIME plays a pivotal role in tumor initiation, progression, and metastatic dissemination.21,22 As a complex immune system network, the TIME consist of tumor-related inflammatory, immune cells, fibroblasts, capillaries, endothelial cells, and extracellular matrix.23,24 Immunotherapy has emerged as a promising treatment for cancer. Unfortunately, some of the patients do not respond to it. Therefore, it is essential to explore the regulatory mechanism of the HCC development and find new targets for immunotherapy.
Studies have shown that tumors with high malignancy expressed high levels of AURKA, suggesting that AURKA upregulation is associated with cancer onset, therapy resistance, tumor recurrence, and metastasis.25,26 Previous studies indicate that AURKA is important in chromosomal alignment and spindle formation during mitosis, and mitotically is more active in cancer cells compared with normal cells. 16 So AURKA expression upregulation in human cancer was not surprising. However, existing AURKA inhibition treatments do not work for all patients with cancer, indicating that many cancers do not fully rely on AURKA for mitosis. 16 Furthermore, in laryngeal squamous cell carcinoma, AURKA may induce epithelial-to-mesenchymal transition via activation of the FAK/PI3K pathway, which is an essential mechanism for tumor survival in HCC. 27
In this study, we first evaluated AURKA expression and clinical relevance using TCGA/GTEx-derived datasets. AURKA was markedly upregulated in HCC at both the mRNA and protein levels. Importantly, elevated AURKA expression was associated with unfavorable survival, supporting its potential prognostic relevance. Consistent with this observation, higher AURKA levels were enriched in more advanced clinicopathological categories, including higher histological grade (G3-G4 vs G1-G2) and more advanced stage and TNM classification, collectively suggesting a relationship between AURKA upregulation and aggressive tumor behavior.28,29
Based on these findings and the ceRNA hypothesis, we explored the specific upstream regulation mechanism of AURKA. Then, we further identified the upstream miRNAs of AURKA, namely hsa-miR-490-3p, by combining expression, prognosis, and correlation analysis with the online database. In this study, we found that hsa-miR-490-3p was downexpressed in HCC tissues and was correlated with a good prognosis in patients with HCC. Previous studies have showed that hsa-miR-490-3p is downregulated in many different tumor entities and exhibits tumor suppressor-like characteristics.30,31 In addition, downregulation of hsa-miR-490-3p by downregulating the Wnt/β-catenin signaling pathway inhibits the progression of lung adenocarcinoma cells. 32 Also, HDAC2 promotes the development and progression of HCC by interacting with the SNHG15/miR-490-3p axis. 33 Long noncoding RNAs can act as the miRNA sponge in a part of ceRNA network, which contribute to the upregulation of targeted genes in HCC,34,35 so we identified the upstream lncRNA for hsa-miR-490-3p, namely DUXAP8, which was upregulated in HCC and associated with poor prognosis.
Previous studies have identified that DUXAP8 as a prognostic biomarker and potential therapeutic target for cancer. 36 We constructed a regulating network based on lncRNA-miRNA-mRNA ceRNA network, which suggests the pathway in which AURKA plays a role in HCC. According to this network, the mechanism of AURKA promotes cell proliferation and tumorigenesis was preliminary illuminated, suggesting the ncRNA effect of AURKA on the emergence and development of HCC (Figure 7).

The model of DUXAP8/ hsa-miR-490-3p/AURKA axis in hepatocarcinogenesis.
Tumorigenesis can be regulated by multiple genes and pathways in various ways.37,38 Compared with single-target antagonists, combining several targets using modern methods may lead to the development of therapeutic strategies and enhance the therapeutic outcomes in cancer treatment. 39 For instance, combinatorial inhibition of AURKA and PLK1 strongly and synergistically reduces tumor progression and cell invasion in diffuse midline gliomas. 40 In various cancer cells, histone deacetylase (HDAC) inhibitors have been demonstrated to inhibit AURKA protein expression. 41 In addition, AURKA inhibitors (AKIs) can downregulate the activity of HDAC proteins, suggesting that adding HDAC inhibitors in combination with AKIs could result in positively synergistic effects. 42
For decades, it has been shown that immune system plays an important role in the development and progression of cancer, and immunotherapy has broadened the breadth and depth of oncology treatment. 43 Earlier studies have revealed that tumor-related immune cell infiltration plays a crucial part in occurrence, progression, and treatment resistance of HCC. As expected, AURKA expression was found to be significantly associated with the TIICs of LIHC. Correlation analysis based on TIMER2.0 database showed a strong positive correlation between AURKA expression and the infiltration levels of neutrophils, macrophages, B cells, dendritic cells, CD8+ T cells, and CD4+ T cells. Therefore, these results confirmed that detecting of the pretreatment level of AURKA may be used to predict the TIICs, which could provide valuable information to guide the choice of immunotherapy for patients with HCC. Some studies have demonstrated that cancer immunotherapy based on immune checkpoint inhibitors can awaken the hosts own immune system to battle against tumor progression. 44 In the analysis of immune checkpoint–related genes, we observed that AURKA expression was positively associated with CTLA4 and LAG3, whereas AURKA expression was negatively associated with CD96, CSF1R, KDR, and PDCD1LG2. This study shows that AURKA may be closely related to the tumorigenesis of HCC and involved in immunoregulatory processes, affecting TIICs and the outcome of immunotherapy. Therefore, targeting AURKA would be a potential therapeutic strategy for the treatment of HCC with high AURKA expression levels.
Although this study provides a comprehensive analysis of the oncogenic role of AURKA in HCC and its regulation through the DUXAP8/hsa-miR-490-3p axis, several limitations must be acknowledged. This study is primarily based on bioinformatics analyses without experimental validation. The regulatory mechanisms identified remain correlative and require further in vitro and in vivo studies. In addition, the lack of clinical immunotherapy data limits the translational applicability of AURKA as a predictive biomarker in HCC. Accordingly, future studies will prioritize experimental validation of the DUXAP8/hsa-miR-490-3p/AURKA regulatory axis in independent HCC cohorts to confirm its biological and clinical relevance. Functional investigations, including gain- and loss-of-function assays and rescue experiments, will be critical to establish causality and elucidate downstream signaling mechanisms. Moreover, integrating single-cell and spatial transcriptomic approaches may provide deeper insights into how this ncRNA-mediated network modulates the TIME. From a translational perspective, expanded interaction-oriented and drug-target–oriented analyses may help uncover therapeutic vulnerabilities within this pathway and guide the development of precision medicine strategies for HCC.
Conclusion
Overall, in this study, we found that AURKA is highly expressed in HCC tissues and is associated with poor prognosis. Furthermore, we proposed a regulatory mechanism of DUXAP8/hsa-miR-490-3p/AURKA axis, which is involved in hepatocarcinogenesis. We have also speculated that the AURKA expression is relative to immune microenvironment and immunotherapy response.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322261435461 – Supplemental material for ncRNA-Mediated Upregulation of AURKA Promotes Hepatocellular Carcinoma Progression and Alters the Immune Microenvironment
Supplemental material, sj-docx-1-bbi-10.1177_11779322261435461 for ncRNA-Mediated Upregulation of AURKA Promotes Hepatocellular Carcinoma Progression and Alters the Immune Microenvironment by Zhitao Chen, Tianshu Chu, Chenchen Ding, Yangjun Gu and Qiyong Li in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
Not applicable.
Author Contributions
All authors contributed substantially to the study conception and design, data acquisition, and data analysis and interpretation. They participated in drafting the manuscript and critically revising it for important intellectual content. All authors approved the final version of the manuscript, agreed to its submission to the current journal, and accept responsibility for the integrity and accuracy of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets used and analyzed in the present study are available from the corresponding authors on reasonable request. The datasets generated and/or analyzed during the current study are available in TCGA (https://portal.gdc.cancer.gov) and GEO (![]() ) database.
) database.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.