
Abstract
The Aurora kinases form a family of 3 genes encoding serine/threonine kinases and are involved in the regulation of cell division during the mitosis. This study was designed to investigate the prognostic role of Aurora kinases in hepatocellular carcinoma (HCC). In this study, we analyzed the expression, overall survival (OS) data, promoter methylation level, and relationship with immunoinhibitors of Aurora kinases in patients with HCC from GEPIA2, UALCAN, OncoLnc, and TISIDB databases. Protein-protein interaction (PPI) network, gene ontology, Kyoto Encyclopedia of Genes and Genomes (KEGG), and Reactome pathway analysis were performed using the STRING database and Cytoscape software. We found that the mRNA expression, stages of HCC, and OS of AURKA and AURKB in HCC tissues were significantly different from control tissues, but there were significant inconsistencies in promoter methylation level and relationship with immunoinhibitors for AURKA and AURKB. None of the above items were significantly different for AURKC. Furthermore, a hub module including AURKA, AURKB, and AURKC was identified within the PPI network constructed with the Molecular Complex Detection (MCODE) plug-in in Cytoscape software. Our results show that AURKB could be a potential biomarker for HCC prognosis.
Introduction
Aurora kinases, which are a family of serine/threonine kinases including Aurora A (AURKA), Aurora B (AURKB), and Aurora C (AURKC), are fundamental kinases in the regulation of cell mitosis, especially in chromosomal segregation. Aurora kinases are also involved in the regulation of meiosis. Aurora kinases deletion may lead to failure of cell division, and disruption of embryonic ontogeny. Overexpression of Aurora kinases has been demonstrated in various cancers, 1 such as breast cancer,2,3 ovarian cancer,4,5 gastric cancer,6,7 and other cancers8-10 and poor prognosis is common for most of them.2,11-13
HCC is one of the most common malignant solid tumors and has the fourth highest mortality worldwide. 14 In 2018, there were approximately 840 000 new cases of HCC and more than 700 000 deaths, with morbidity and mortality rates continuing to increase for both men and women. 15 Although treatments such as chemotherapy, radiotherapy, and other treatments have achieved some inspiring effects, the prognosis remains poor due to the high recurrence rate. 16 This situation urged researchers to focus on the prognosis of HCC. Furthermore, effective predictive markers to evaluate the prognosis of HCC patients are urgently needed.
The relationship between the abnormal expression of Aurora kinases and clinical HCC patient prognosis has been occasionally reported.17,18 To the best of our knowledge, bioinformatics analysis has not yet been applied to explore the role of Aurora kinases in HCC. Next generation sequencing (NGS) has changed dramatically in recent years. Based on data from the online databases, we studied the expression of Aurora kinases and their prognostic value in HCC.
Materials and Methods
Database
GEPIA is a novel interactive-based web server that is mainly designed for dealing with the RNA sequencing expression data, including data from more than 9000 tumors and 8000 normal samples from The Cancer Genome Atlas (TCGA) and GTEx projects. GEPIA provides customizable analysis, such as tumor/normal differential expression analysis, transcripts per million (TPM) analysis, pathological stage analysis, patient survival analysis, and so on. 19 GEPIA2 is an updated and enhanced version of GEPIA.
UALCAN is a comprehensive, user-friendly, and interactive web resource for analyzing cancer OMICS data (from TCGA and MET500). 20 UALCAN is built on PERL-CGI with high-quality graphics using JavaScript and CSS.
TISIDB is a web portal for assessing tumor and immune system, which integrates multiple heterogeneous data types: genomics, transcriptomics, and clinical data of 30 cancer types from TCGA. 21
OncoLnc is a newly available resource for Cox coefficient analysis and utilizes data from 21 cancers in TCGA. 22 Genes are first ranked in each cancer by their Cox coefficient, and these ranks are then combined by applying Fisher’s method.
DNMIVD is a comprehensive annotation and interactive visualization database for assessing the DNA methylation profiles of diverse human cancers constructed with high-throughput microarray data from the TCGA and Gene Expression Omnibus (GEO) databases.
Immunoinhibitor analysis
Immunoinhibitors play a vital role in the progression of tumors, and are AURKA/AURKB expression related to immunoinhibitors in HCC, and which immunoinhibitors are our target? We examined which immunomodulators might be regulated by AURKA/AURKB on TISIDB. Spearman’s test was used to measure the correlation between AURKA/AURKB and immunoinhibitors.
GO and KEGG pathway analysis of the Aurora kinase module
Gene ontology (GO) analysis divides the functions of genes into 3 parts: cellular component (CC), biological pathway (BP), and molecular function (MF). The Kyoto Encyclopedia of Genes and Genomes (KEGG) is an online database for systematic analysis of gene function and genomic information, that helps researchers to study gene expression information from a network perspective. Reactome pathway analysis provides molecular details of signal transduction, transport, metabolism, and other cellular processes in the form of an ordered network of molecular metabolic maps.
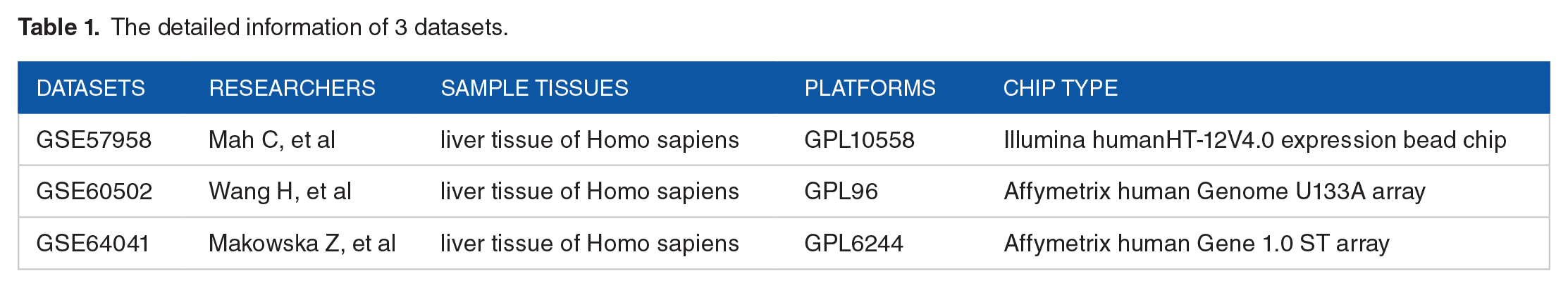
First, the online database STRING 23 was employed to identify the differentially expressed genes (DEGs) in 3 datasets (GSE57958, GSE60502, GSE64041) (Table 1) from the GEO database, these DEGs and the Aurora kinase genes were used to construct the PPI network. Second, Cytoscape software 24 was utilized to analyze the interactions between all the above-mentioned genes in HCC.
The detailed information of 3 datasets.
Statistical analysis
The correlation between the expression levels of Aurora kinases and patient OS was analyzed using the Kaplan-Meier method. Spearman’s rank correlation coefficient was used for the immune analysis between AURKA/AURKB and immunoinhibitors. Statistical analysis was performed with SPSS 20.0 software. Significant difference was established at P < 0.05.
Results
Expression levels of Aurora kinase genes in HCC
By comparing 160 normal liver tissues and 369 HCC tissues with the GEPIA2 database, it was found that the mRNA expression levels of AURKA and AURKB were significantly higher in patients with HCC than in controls (P < 0.05), while AURKC was not significantly different between the HCC group and the control group (P > 0.05) (Figure 1). In addition, we confirmed the above results with the UALCAN database (Figure 1).

The mRNA expression level between HCC tumor and normal tissues from GEPIA 2 (a-c). (a) AURKA (P < 0.05) (b) AURKB (P < 0.05) (c) AURKC (P > 0.05). The mRNA expression level between HCC tumor and normal tissues from UALCAN (d-f). (d) AURKA (P < 0.05) (e) AURKB (P < 0.05) (f) AURKC (P > 0.05). TPM means transcripts per million, LIHC means liver hepatocellular carcinoma.
Correlation between expression of Aurora kinase genes and tumor stage in HCC
Based on the above analysis, the correlation between Aurora kinase gene expression and tumor stage in HCC were was analyzed by GEPIA2. The results demonstrated that there was a significant correlation between the expression levels of AURKA/AURKB and the tumor stage in HCC(P < 0.05), while AURKC was not significantly correlated with tumor stage (P > 0.05) (Figure 2). We further confirmed the above results by UALCAN and TISIDB databases (Figure 2).

The relationship between gene expression level and HCC stages (I, II, III, IV) from GEPIA 2 (a-c). (a) AURKA (P < 0.05) (b) AURKB (P < 0.05) (c) AURKC (P > 0.05). The relationship between gene expression level and HCC stages (I, II, III, IV) from UALCAN (d-f). (d) AURKA (P < 0.05) (e) AURKB (P < 0.05) (f) AURKC (P > 0.05). The relationship between gene expression level and HCC stages (I, II, III, IV) from TISIDB (g-i). (g) AURKA (P < 0.05) (h) AURKB (P < 0.05) (i) AURKC (P > 0.05).
Correlation between Aurora kinase gene expression and OS in HCC
We further explored the vital role of Aurora kinases in the survival of patients with HCC. Kaplan-Meier Plotter methods were used to analyze the correlation between the mRNA levels of Aurora kinases and the OS of patients with HCC by using GEPIA2, OncoLnc, and TISIDB. The Kaplan-Meier curve and log-rank test analysis showed that the AURKA and AURKB mRNA levels were significantly correlated with the OS of patients with HCC (P < 0.05), while there was no significant correlation for AURKC (P > 0.05) (Figure 3). The patients with high mRNA levels of AURKA and AURKB were predicted to have a worse prognosis.

The relationship between gene expression level and overall survival in HCC from GEPIA 2 (a-c). (a) AURKA (P < 0.05) (b) ALRKB (P < 0.05) (c) AURKC (P > 0.05). The relationship between gene expression level and overall survival in HCC OncoLnc (d-f). (d) AURKA (P < 0.05) (e) AURKB (P < 0.05) (f) AURKC (P > 0.05). The relationship between gene expression level and overall survival in HCC from TISIDB (g-i) AURKA (P < 0.05) (h) AURKB (P < 0.05) (i) AURKC (P > 0.05). The red line represents the high gene expression group and the blue line the indicates the low gene expression group.
Spearman correlation between AURKA/AURKB and immunoinhibitors
After determining the prognostic role of AURKA and AURKB, we performed Spearman correlation analysis between AURKA/AURKB and immunoinhibitors for HCC (Figures 4 and 5). The Spearman correlation showed that elevated AURKB was significantly correlated with PDCD1 (rho = 0.326, P < 0.05) and CTLA-4 (rho = 0.349, P < 0.05) (Figure 6), but elevated AURKA was only significantly correlated with CTLA-4 (rho = 0.159, P < 0.05), and not significantly correlated with PDCD1 (rho = 0.069, P > 0.05) (Figure 6).

The spearman correlations between expression of AURKA and immunoinhibitors. The column labeled with red arrow is LIHC which is wanted. The red color means positive correlation, the blue color means negative correlation.

The spearman correlations between expression of AURKB and immunoinhibitors. The column labeled with red arrow is LIHC which is wanted. The red color means positive correlation, the blue color means negative correlation.

The spearman correlations between expression of AURKA/AURKB and PDCD 1/CTLA-4. (a) the spearman correlation between expression of AURKA and PDCD1 (P > 0.05) (b) the spearman correlation between expression of AURKA and CTLA-4 (P < 0.05) (c) the spearman correlation between expression of AURKB and PDCD1 (P < 0.05) (d) the spearman correlation between expression of AURKB and CTLA-4 (P < 0.05). rho means the spearman correlation value. The closer rho gets to 1, the greater the correlation.
PPI network of Aurora kinases genes and DEGs
To identify the PPI of Aurora kinase genes in HCC, the STRING was employed to construct a PPI using 86 DEGs from 3 GEO expression datasets (Table 1) and Aurora kinase genes. Then, Cytoscape software was utilized to visualize the PPI network and analyze the interactions between the DEGs and Aurora kinase genes in HCC. The network analyzer plug-in MCODE was used to identify the hub module of the PPI (Figure 7).

The protein-protein interaction (PPI) network of AURKA/AURKB/AURKC and DEGs by STRING. An important module (marked yellow) from the PPI network was found using MCODE.
The PPI network contained 88 nodes and 189 edges, the average node degree was 4.3, and the local clustering coefficient was 0.555 (PPI enrichment P < 1.0E-16). We found one significant module from the PPI network for further analysis using MCODE. In this module, the network score was 10.6, including 11 genes (AURKA, AURKB, AURKC, CDKN3, CCNB2, NCAPG, ASPM, NUSAP1, TOP2A, PRC1, and MELK). Finally, the 11 key genes were subjected to get the BP, MF, CC, KEGG pathway, and Reactome pathway analysis by the STRING (Figure 8).

GO analysis, KEGG, and Reactome Pathways of the module from Figure 7: (a) biological process of GO analysis, (b) molecular function of GO analysis, (c) cellular component of GO analysis, (d) KEGG pathway analysis, and (e) Reactome pathway analysis.
Promoter methylation level of Aurora kinases and Spearman correlation between promoter methylation and gene expression
It is well-known that DNA methylation is negatively correlated with gene expression.25,26 By analyzing the DNMIVD database, we observed that the AURKB promoter methylation level was significantly lower in HCC than in control samples (P < 0.05) and was negatively correlated with the expression of AURKB (P < 0.05) (Figure 9), providing further support for the high expression of AURKB in HCC. As expected, the AURKC promoter methylation level was not significantly different between HCC and control samples (P > 0.05) and was not correlated to its expression (P > 0.05) (Figure 9). However, AURKA showed unexpected results: the AURKA promoter methylation level was not significantly different between HCC and control samples (P > 0.05) and was positively correlated with the expression of AURKA (P < 0.05) (Figure 9).

Promoter methylation level of AURKA/AURKB/AURKC between tumor and normal tissue by DNMIVD. (a) AURKA (P > 0.05) (b) AURKB (P < 0.05) (c) AURKC (P > 0.05). The spearman correlation between FPKM and promoter methylation of AURKA/AURKB/AURKC. (d) AURKA (positive correlation) (e) AURKB (negative correlation) (f) AURKC (negative correlation). FPKM represents expression level.
Discussion
The dysregulation of Aurora kinase genes has been implicated in many cancers,27-30 including gastric cancer, ovarian cancer, hepatocellular carcinoma, colon cancer, and so on.
Although the effects of Aurora kinases on the prognosis of some cancers have been elucidated, further bioinformatics analysis of HCC should be carried out. To the best of our knowledge, we present the first study of the mRNA expression and prognostic value of all the Aurora kinase family members in patients with HCC. We sincerely hope that this bioinformatics analysis will provide evidence to further understanding of the prognosis of patients with HCC.
Aurora kinases have a highly conserved structure, and are composed of 3 homologous parts, an N-terminal domain, a protein kinase domain and a C-terminal domain.31,32 Each Aurora kinase is activated by autophosphorylation of specific T-loop residues: Thr288 (AURKA), Thr232 (AURKB), and Thr195 (AURKC). Aurora kinases lose function after dephosphorylation mediated by protein phosphatase 1. 33
AURKA initially localizes to the duplicate centrosome at the S phase and transfers to the bipolar spindle microtubules during the medium-term of mitosis, finally shifts to the perinuclear materials of the duplicate cells at the telophase of mitosis. 34 Studies have shown that AURKA is distributed outside of the nucleus in cancer cells 35 and participates in tumorigenesis through a variety of mechanisms. AURKA can directly interact and phosphorylate the glycolytic enzyme LDHB, which promotes tumor progression after a series of biochemical reactions. 36 In addition, lncRNA TUG1 positively upregulates the expression of AURKA, thus promotes cancer cell proliferation, migration, and invasion. 37 In this study, the GEPIA2 and UALCAN databases demonstrated that the expression of AURKA was higher in HCC than in control tissues. Furthermore, the AURKA expression was significantly correlated with tumor stage in patients with HCC. We also revealed that high AURKA expression was significantly correlated with less OS by GEPIA2, OncoLnc, and TISIDB data. Metastasis mediated by epithelial-mesenchymal transition (EMT) is a very important cause of poor prognosis in cancer patients, and AURKA can upregulate the expression of vital EMT transcription factor SLUG. 38 In addition, AURKA also promotes EMT by activating WNT/AKT signaling pathways and simultaneously reduces promoter site H3K4/H3K27 methylation. 39 Originally, we expected to confirm the expression of AURKA by its promoter methylation level, but AURKA promoter methylation level was not significantly different in HCC from normal tissues, moreover, generally speaking, methylation level is negatively correlated with gene expression, even if AURKA promoter methylation level was not significantly different, AURKA promoter methylation level should have been negatively correlated with expression of AURKA, but really it was positively correlated with expression in Figure 9. Therefore, considering unreasonable contradictions for AURKA, the role of AURKA in HCC prognosis should be in dispute and requires further studies.
AURKB expression emerges at early G2 phase and mainly localizes to the centromere in prometaphase and metaphase, and the central spindle in anaphase. 40 AURKB is able to activate the expression of CCND1, which is a direct downstream target of AURKB, and plays a key role in gastric cancer cell proliferation. 41 Furthermore, it has been shown that AURKB plays an important role in the regulation of lymphoma cell survival and proliferation by activating the AKT/mTOR signaling pathway. 42 In this study, we demonstrated that the expression of AURKB was higher in HCC than in control tissues with data from GEPIA2 and UALCAN databases, and AURKB expression was significantly correlated with tumor stage in patients with HCC. Then, we determined the prognostic value of AURKB in patients with HCC, and high AURKB expression was significantly correlated with poor OS according to GEPIA2, OncoLnc, and TISIDB analysis. Consistently, the AURKB promoter methylation level was significantly lower in HCC than in control tissues.
Few studies of AURKC have been published. Recent studies have revealed that AURKC is primarily expressed in the interphase and combines with chromosomes to play a role during mitosis. 43 However, the exact distribution and translocation of AURKC during mitosis have not been determined. In contrast to AURKA and AURKB, AURKC is expressed specifically in mammalian testes rather than in other somatic tissues. The role of AURKC in promoting tumor progression is probably due to its overlapping and complementary functions with AURKB. 44 Although the mechanism is still unclear, we found that it was not significantly different in HCC from control tissues in all aspects including the expression of RNA, tumor stage, prognostic value, and promoter methylation level of AURKC in this study. All the results revealed that AURKC is not a prognostic indicator for HCC.
Programed cell death protein 1 (PD1 or PDCD1), also known as CD279, is the key regulatory factor in the effector phase of T cell-mediated immune responses, and is expressed on activated T lymphocytes, and natural killer (NK) cells, in particular, it is highly expressed on tumor-specific T cells. 45 Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an inhibitory receptor belonging to the CD28 subfamily, and it is a crucial inhibitor of T cell proliferation and function.46,47 Tumors can efficiently suppress immune responses by activating negative regulatory pathways, which are named immune checkpoints (ICPs). 47 PD1 and CTLA-4 are good choice for tumor in the ICP. It has been reported that effective antitumor immune surveillance in the liver microenvironment is impaired, especially when the PD1/PDL1 signaling pathway is greatly involved in this process. 48 The overexpression of CTLA-4 in HCC leads to uncontrolled growth of the tumors. 49 In recent years, increasing attention has been given to the role of immune regulation in tumors. From the results of Spearman correlation analysis between AURKA/AURKB and immunoinhibitors of HCC, AURKB might have a synergistic effect on the immunosuppression of T lymphocytes in the liver immune environment, which promotes the progression of the tumor. The liver is a tolerogenic organ, therefore, the problem of immunosuppression in hepatocellular carcinoma is more complex, and the mechanism needs further research on AURKB and immunoinhibitors.
The GO analysis of the PPI module containing 11 genes, including AURKA, AURKB, and AURKC, showed that the enriched BP terms was mainly associated with the cell cycle process, mitotic cell cycle process, and cell division. The enriched CC terms mainly included chromosome passenger complex, midbody, and spindle microtubules. The enriched MF terms mainly included the histone serine kinase activity and protein serine/threonine/tyrosine kinase activity. The GO analysis results were highly consistent with the molecular role of Aurora kinases. SUMOylation of DNA replication proteins was the most enriched term in the Reactome pathway analysis.
Conclusions
In this study, we systemically analyzed the expression and prognostic value of Aurora kinases in HCC by virtue of bioinformatic analysis based on data from several platforms. Although there was a significant expression difference for AURKA, which was significantly correlated with the stages of HCC, and high AURKA expression was correlated with reduced OS in HCC, but its promoter methylation level and relationship with immunoinhibitors showed contradictions, so more research needs to be performed to demonstrate the mechanism of AURKA. Our work indicates that AURKB could serve as a potential prognostic marker for HCC. We hope our work will enrich the understanding of the prognostic value of AURKB for patients with HCC.
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Key Research and Development Program of Hainan Province (No. ZDYF2018174), the Major Science and Technology Program of Hainan Province (No. ZDKJ2017007), and the Project of Health Department of Hainan Province (NO. 19A200156). We sincerely thank TCGA, GEPIA2, UALCAN, TISIDB, OncoLnc, DNMIVD, Cytoscape, STRING, and GEO databases for their data sharing.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Data collection and curation, Jingchuan Xiao; Data analysis, Jingchuan Xiao; Writing-original draft and visualization, Jingchuan Xiao; Funding acquisition, Yingai Zhang; Writing-review & editing, Yingai Zhang.
Data Availability
The figure and data used in this study are available from the public database with reasonable request.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.