
Abstract
As a groundbreaking advancement in vaccinology, messenger RNA (mRNA) vaccines have transformed the field by offering rapid, flexible, and scalable solutions for combating infectious diseases. However, the efficacy, stability, and immunogenicity of mRNA vaccines are highly dependent on the optimization of their sequences. Recent progress in synthetic biology and computational methods has enabled the optimization of mRNA sequences to enhance their properties, holding the promise to provide deeper insights into the design principles of effective mRNA vaccines. However, it remains a major challenge to determine how to best optimize mRNA sequences for diverse biological contexts and therapeutic applications. In this review, we provide an in-depth analysis of the current advancements in optimizing mRNA vaccine sequences, put forward a comprehensive overview of the latest computational and biological approaches in this field, with a particular focus on the biological mechanisms underlying mRNA translation efficiency and stability, highlighting several quantitative indicators that may affect vaccines’ performance, and summarize some methods to optimize mRNA vaccine by algorithms. We also propose the limitations of current models and the need for further research to address the complexity of biological systems.
Introduction
The advent of messenger RNA (mRNA) vaccines has revolutionized the field of medical therapeutics, offering a versatile and rapidly deployable solution for addressing a wide range of diseases. mRNA vaccines function by delivering genetic instructions to host cells, prompting the production of specific antigens that elicit a targeted immune response. 1 This innovative approach has garnered significant attention due to its flexibility, scalability, and rapid development timeline. 2 The successful deployment of COVID-19 mRNA vaccines has demonstrated the immense potential of this technology, highlighting its ability to rapidly respond to emerging pathogens. Beyond infectious diseases, mRNA vaccines are also being explored for cancer immunotherapy, personalized medicine, and the treatment of chronic conditions.3,4 Their capability to induce robust humoral and cellular immune responses makes mRNA a powerful tool in both prophylactic vaccine and therapeutic vaccine development. However, the efficacy and stability of mRNA vaccines are highly dependent on the optimization of mRNA sequences, which directly impacts antigen production and immune response. 5
Recent advancements in sequence optimization have markedly enhanced the performance of mRNA vaccines. The untranslated regions (UTRs) and coding sequence (CDS) are critical elements that regulate translation efficiency (TE) and mRNA stability. 6 Advances in understanding the biological mechanisms underlying mRNA translation and stability have led to the development of quantitative metrics such as mean ribosome load (MRL), codon adaptation index (CAI), and GC content. These metrics provide valuable insights into optimizing mRNA sequences for enhanced performance. For instance, optimizing the 5′UTR sequence can significantly enhance MRL, a key metric for TE. In addition, chemical modified nucleotide such as pseudouridine (Ψ) and 1-methyl-pseudouridine (m1Ψ) have been shown to improve mRNA TE and stability and reduce immunogenicity. 7 The nucleotide modifications, along with optimized codon usage and secondary-structure refinement, have been demonstrated to enhance protein expression and overall vaccine efficacy. Despite these achievements, the complexity of biological systems and the variability in cellular environments present ongoing challenges for sequence optimization.
Concurrently, the rapid advancement of artificial intelligence (AI) and machine learning (ML) has garnered significant attention, offering new solutions to address these challenges and transforming numerous fields, which includes biotechnology and medicine. 8 AI algorithms, particularly deep learning models, have shown remarkable capabilities in handling complex biological data and predicting outcomes with high accuracy. Convolutional neural networks (CNNs) and recurrent neural networks (RNNs) have been particularly effective in analyzing large datasets and identifying patterns that are not easily perceptible through traditional methods. 3 The ability of these algorithms to process and integrate vast amounts of data has made them invaluable tools for understanding and optimizing biological sequences in biologically meaningful manner. In the context of mRNA vaccines, AI algorithms can predict ribosome loading, mRNA stability, and TE, thereby facilitating the design of mRNA sequences with improved performance. The integration of AI with experimental biology has opened new avenues for optimizing mRNA vaccines, offering a more systematic and efficient approach to sequence design. 9
These theoretical advancements have been concretely validated by AI models, greatly showing that the integrative potential of AI in optimizing biological systems remains vast. AI models, such as Optimus 5-Prime, have demonstrated the ability to accurately predict ribosome loading and mRNA stability, outperforming traditional optimization methods. 10 The use of AI algorithms offers several advantages, including the ability to handle complex biological systems, identify subtle sequence variations, and provide rapid and accurate predictions. 8 In addition, AI models can be trained on diverse datasets, enabling them to generalize across different cell types and condition. This flexibility is crucial for developing mRNA vaccines that are effective in various biological contexts.
This review aims to provide a comprehensive summary of the recent progress in sequence optimization of mRNA vaccines, focusing on the integration of AI algorithms with biological mechanisms and quantitative metrics. We have reviewed numerous studies that utilize AI models to facilitate mRNA sequences design or optimization, highlighting their achievements and limitations. By summarizing and synthesizing these studies, we aim to provide a clear understanding of the current state of the field and identify future directions for research. This review underscores the importance of AI in optimizing mRNA vaccine sequences and highlights the potential of these algorithms in enhancing vaccine efficacy and stability. We hope that this comprehensive analysis will serve as a valuable resource for researchers and developers working in the field of mRNA vaccines, guiding future efforts in sequence optimization and vaccine design.
Biological Mechanisms
There are several biological mechanisms that can potentially affect the effectiveness of mRNA vaccine. And they are discussed in the following sections:
Untranslated regions
UTRs are non-CDS flanking the protein-coding region of mRNA and are integral to post-transcriptional regulation. 11 Located at the 5′ and 3′ ends of the mRNA, these regions do not encode amino acids but contain critical regulatory elements that govern TE, mRNA stability, and subcellular localization. The 5′ UTR, preceding the start codon, facilitates ribosome recruitment and modulates translation initiation through secondary structures or upstream open reading frames (uORFs). The 3′ UTR, following the stop codon, harbors motifs such as microRNA (miRNA) binding sites, AU-rich elements (AREs), and polyadenylation signals, which influence mRNA decay, translational repression, and interactions with RNA-binding proteins. UTRs thus act as dynamic platforms for coordinating gene expression in response to cellular signals, developmental cues, or stress, underscoring their importance in both normal physiology and disease. Computational optimization of UTRs integrates sequence and structure features—such as local secondary structure, start-codon accessibility, upstream AUGs and uORFs, known regulatory motifs, and RNA–protein binding predictions—using thermodynamic models and machine-learning approaches to design UTR variants that maximize TE and stability under defined cellular contexts while avoiding inhibitory elements.
Upstream ORF
uORFs are short sequences in mRNA 5′UTRs that transiently initiate translation, primarily suppressing main ORF expression by sequestering ribosomes or inducing premature termination. 12 They reduce TE by 30%–80%, depending on Kozak sequence strength, uORF length (optimal ⩽ 30 codons for ribosome reinitiation), and termination codon proximity. Under stress, certain uORFs enhance translation. Plant uORFs often trigger nonsense-mediated decay (NMD), while human uORFs rarely activate this pathway. Optimization of uORFs involves identifying and scoring uORF features using tools like uORFscan and then redesigning the 5′ UTR to remove inhibitory uORFs, weaken their initiation signals, or engineer conditional uORFs.
Translation initiation factor binding
Translation eukaryotic initiation factors (eIFs) are pivotal for ribosome recruitment and decoding mRNA into proteins. 13 The eIF4F complex—comprising eIF4E (cap-binding protein), eIF4A (RNA helicase), and eIF4G (scaffold protein)—binds the 5′ cap (m7G) of mRNA to unwind secondary structures and assemble the 48S preinitiation complex. eIF4E specifically recognizes the cap structure, while eIF4A resolves inhibitory RNA folds in the 5′UTR to facilitate ribosome scanning. Poly(A)-binding proteins (PABPs) at the 3′ tail further enhance initiation by bridging eIF4G and forming a closed-loop mRNA structure, synergistically boosting TE. Computational optimization targets these determinants by modeling eIF–mRNA interactions using features such as start-codon accessibility, local secondary-structure stability, Kozak context strength, eIF4F recruitment propensity, and scanning barrier scores, and then redesigning the 5′ UTR and proximal CDS to maximize predicted initiation efficiency while avoiding inhibitory structures or competing elements such as strong uORFs.
5′ terminal oligopyrimidine tract (5′TOP)
The 5′TOP is a critical regulatory element found in the 5′ untranslated regions (5′ UTRs) of mRNAs encoding ribosomal proteins and translation factors. 14 This motif, characterized by a cytosine at position + 1 followed by a sequence of 4 to 15 pyrimidines, plays a pivotal role in translational control, particularly in response to cellular growth signals and nutrient availability. The 5′TOP motif is known to mediate growth-dependent translational regulation, with its activity modulated by pathways such as the mechanistic target of rapamycin (mTOR) signaling cascade, which is sensitive to rapamycin inhibition. In mRNA design, optimization of the 5′TOP feature involves computational tuning of the length, pyrimidine composition, and structural context of the 5′ terminal motif, combined with modeling of mTOR responsiveness, cap-proximal secondary structure, and ribosome accessibility, to either enhance conditional translational control or deliberately avoid TOP-like elements for constitutive expression, depending on the therapeutic or experimental objective.
Kozak sequence
The Kozak sequence is a conserved nucleotide motif (5′-GCC(A/G)CCAUGG-3′) flanking the start codon (AUG) in eukaryotic mRNA, critical for efficient translation initiation. 15 Discovered by Marilyn Kozak in the 1980s, its −3 position (A/G) and +4 position (G) are key determinants: a purine at −3 enhances ribosomal recognition by 4-fold, while a pyrimidine (C/T) reduces initiation efficiency and increases sensitivity to adjacent sequence variations. This sequence acts as a ribosomal “landing pad,” directing the 40S subunit to unwind 5′UTR secondary structures via eIF4A helicase activity and position AUG in the ribosomal P-site for accurate methionine-tRNA loading. Optimization of the Kozak sequence involves engineering Kozak variants that maximize TE without introducing unintended upstream initiation.
MicroRNA binding
miRNAs are short non-coding RNAs (~22 nucleotides) that post-transcriptionally regulate gene expression by binding to complementary sequences in target mRNA 3′ untranslated regions (3′UTRs), triggering translational repression or mRNA degradation. 16 This interaction relies on a “seed sequence” (nucleotides 2–8 of the miRNA) pairing with mRNA targets, often amplified by auxiliary binding sites in coding regions. For example, miR-155 and miR-150 modulate immune responses by targeting transcripts encoding cytokines (eg, IL-12) and co-stimulatory molecules (eg, CD40) in dendritic cells (DCs). In mRNA vaccine design, optimization involves computational identification and elimination of high-affinity miRNA binding sites, evaluation of site accessibility using RNA folding models, and incorporation of synonymous mutations or UTR redesign to reduce unintended miRNA interactions, thereby enhancing stability and translation while preserving protein-CDS.
CDS: sequence and structure
The CDS is the central functional region of mRNA, spanning from the start codon (AUG) to the stop codon, and directly determines the amino acid sequence of the translated protein. 17 Beyond this primary role, the nucleotide composition and structural features of the CDS itself are critical determinants of mRNA stability, TE, and even immunogenicity. Optimization of the CDS is therefore a cornerstone of mRNA vaccine design. Computational optimization of the CDS typically involves synonymous recoding to balance codon usage and RNA structure, using metrics such as CAI/tAI, minimum free energy (MFE) or base-pairing probability profiles, codon pair bias, and ribosome accessibility windows, often guided by multi-objective optimization or machine-learning models to maximize TE while preserving protein sequence and regulatory constraints.
Preferred codon usage
Codon usage which refers to the choice of codon is a key factor in optimizing mRNA sequences. 18 Codons that are more frequently used in the target organism can enhance TE without altering the protein sequence. For example, replacing rarely used codons with more frequently occurring ones can increase protein expression levels. This optimization strategy helps ensure that the antigen is produced at optimal levels, which is critical for eliciting a strong immune response. However, it must be used judiciously, as some rare codons may be necessary for proper protein folding. Codon usage optimization is quantitatively described using CAI, tAI, relative synonymous codon usage (RSCU), codon pair bias scores, codon harmonization metrics, and host-specific codon frequency distributions that are directly computable from sequence data.
GC content
The percentage of guanine (G) and cytosine (C) nucleotides in a nucleic acid sequence—profoundly influences mRNA stability, secondary structure, and translational efficiency. 18 Elevated GC content (>50%) strengthens mRNA stability through enhanced base-pairing (three hydrogen bonds for G:C vs two for A:T), reducing degradation by nucleases. However, excessive GC-rich regions (>70%) risk forming stable secondary structures (eg, hairpins), which impede ribosome scanning and reduce TE. Uracil (U) is often replaced with guanine (G) or cytosine (C) to reduce the likelihood of mRNA degradation by nucleases. In addition, optimizing AGC content can help in reducing the immunogenicity of the mRNA, which is important for minimizing adverse immune responses. Computational optimization of GC content is performed by recoding synonymous codons to tune local and global GC levels while monitoring other quantitative metrics, enabling systematic identification of GC profiles that maximize translational output without inducing excessive secondary structure.
Secondary structure
The secondary structure of mRNA refers to the specific folding patterns formed by the mRNA molecule through intramolecular base-pairing interactions. 19 These structures, such as hairpin loops and stem-loops, are stabilized by hydrogen bonds between complementary nucleotides. More stable global structures can protect the mRNA from enzymatic degradation by ribonucleases, thereby increasing its intracellular half-life. However, strong stem-loops or hairpins within CDS can cause ribosomes to pause or even dissociate during elongation, leading to truncated protein products or reduced overall protein synthesis. Optimization of secondary structure focuses on structure-aware metrics—such as MFE, base-pairing probability, accessibility of the 5′ UTR and start codon, and local folding profiles—often optimized in a position-specific and windowed manner rather than globally.
G-quadruplex
G-quadruplexes (G4s) are non-canonical nucleic acid structures formed by guanine-rich sequences, where four guanines arrange into planar tetrads stabilized by Hoogsteen hydrogen bonds and monovalent cations (eg, K+ or Na+). 20 These structures exhibit functional duality: in mRNA 5′UTRs, they often act as translational roadblocks by stalling ribosome scanning. Computational tools can be used to predict and eliminate potential G-quadruplex-forming sequences, thereby optimizing the mRNA sequence for enhanced TE and stability.
Chemical modification
Chemical modifications of mRNA are pivotal for enhancing stability, reducing immunogenicity, and improving translational efficiency. 21 Unmodified ssRNA and dsRNA contaminants in IVT mRNA can be recognized by cellular pattern-recognition receptors (PRRs) like Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), and protein kinase R (PKR), triggering the production of type I interferons and other pro-inflammatory cytokines. Key strategies include nucleotide substitutions (eg, pseudouridine, N1-methylpseudouridine (m1Ψ), 5-methylcytidine (m5 C)), which minimize recognition by innate immune sensors and suppress interferon responses. Chemical modifications in mRNA (such as pseudouridine, N1-methylpseudouridine, m6A, or 5-methylcytidine) alter base-pairing energetics, stacking interactions, and local flexibility, thereby reshaping secondary structure by stabilizing or destabilizing specific stems and loops and changing folding kinetics. As a result, modified mRNAs often adopt structural ensembles that differ from their unmodified counterparts. Secondary-structure prediction for chemically modified mRNA is therefore difficult because most algorithms and energy models are trained on unmodified nucleotides, lack modification-specific free-energy parameters, and cannot accurately account for heterogeneous, position-specific modifications or altered co-transcriptional folding behavior.
Internal ribosome entry sites
Internal ribosome entry sites (IRESs) are structured RNA elements in mRNA that enable cap-independent translation initiation. 22 IRES elements can directly recruit the 40S ribosomal subunit to an internal location on the mRNA, via conserved secondary/tertiary structures and interactions with trans-acting factors like polypyrimidine tract-binding protein (PTB) or eIFs, often in close proximity to the translation start codon. IRESs are crucial for the translation of circular RNAs (circRNAs), which inherently lack the 5′ cap structure required for canonical, cap-dependent translation initiation. IRES function is tightly coupled to higher-order RNA secondary and tertiary structures and to specific sequence–structure motifs that control ribosome landing, initiation efficiency, and cell-type specificity.
The Biological mechanisms affecting mRNA vaccine design are summarized in Figure 1.
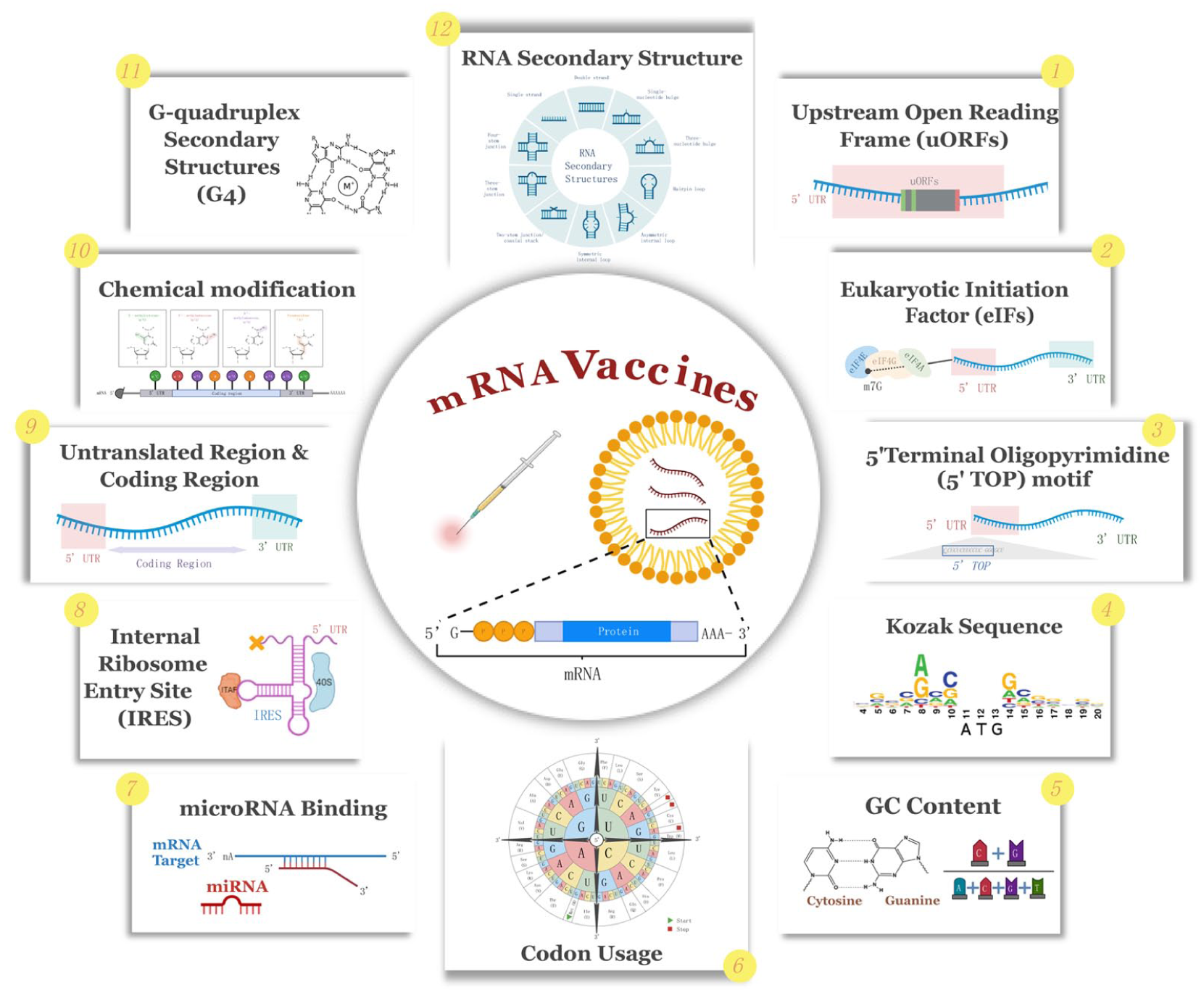
Biological mechanisms affecting mRNA vaccine design.
Quantitative Metrics
The design and refinement of mRNA sequences for vaccines and therapeutics rely heavily on quantitative metrics that serve as proxies for desirable biological outcomes. These metrics allow computational models to evaluate and compare different sequence candidates, guiding the optimization process toward enhanced stability, translational efficiency, and reduced immunogenicity. Understanding these metrics is crucial for interpreting the outputs of design algorithms and for appreciating the multifaceted nature of mRNA optimization.
Minimum free energy
MFE is a thermodynamic measure representing the stability of the most stable secondary structure an RNA molecule is predicted to adopt. It is expressed in kcal/mol, with a lower (more negative) MFE value indicating a more stable and more extensively base-paired structure, as more energy would be required to unfold it. The principle underlying MFE-based optimization is that more stable overall mRNA structures are generally more resistant to enzymatic degradation within the cell, thus correlating with increased mRNA half-life. 23 Consequently, MFE is a primary target for many structural optimization algorithms, such as CDSfold 24 MFE is typically calculated using dynamic programming algorithms, like those implemented in widely used software packages such as RNAfold (part of the ViennaRNA package) or mfold, which employ nearest-neighbor thermodynamic parameters to estimate the energy contributions of various structural motifs (eg, stems, loops, bulges). 25
While a lower MFE is often sought, it is important to recognize its limitations. MFE represents a global property of the most stable predicted structure and does not fully capture the dynamic ensemble of structures an RNA molecule might adopt in vivo. Furthermore, excessive structural stability, particularly in regions critical for translation (like the start codon or ribosome binding site), can actually impede ribosome scanning or elongation, thereby reducing protein expression. Overly stable, long dsRNA regions can also trigger innate immune responses. Thus, MFE optimization must be balanced with other functional considerations. 26
Codon Adaptation Index
The CAI is a widely used metric to quantify the extent to which the codon usage in a given gene conforms to the codon usage bias observed in a reference set of highly expressed genes from a specific organism or cell type. 27 The CAI value ranges from 0 to 1, where a value of 1.0 indicates that the gene exclusively uses the most frequently occurring synonymous codons for each amino acid. CAl is calculated using the following equation:

where

where f and
Codon optimization based on maximizing CAI has been a standard strategy in recombinant protein production and mRNA therapeutic design. 29 However, solely maximizing CAI can have drawbacks. It may lead to the creation of unintended RNA secondary structures or alter GC content suboptimally. Moreover, extremely rapid translation might compromise the correct co-translational folding of some proteins. 30 In addition, standard CAI calculations often use genome-wide codon usage tables, which may not reflect tissue-specific or condition-dependent variations in tRNA availability. 31 Therefore, CAI is often used as one of several objectives in multi-parameter optimization algorithms.
Mean ribosome load
MRL is a measure of the average number of ribosomes actively translating a given mRNA molecule at a specific point in time. It is often determined experimentally using techniques like ribosome profiling (Ribo-seq), which involves sequencing the mRNA fragments protected by ribosome. 32 A higher MRL generally indicates a higher rate of translation initiation and/or efficient elongation, leading to increased protein synthesis from that mRNA species. MRL can serve as a more direct readout of translational activity than purely sequence-derived metrics like CAI. Computational models, such as the UTR-LM, are being developed to predict MRL based on sequence features, particularly those within the 5′ UTR, thereby providing an indirect quantification of TE to guide UTR optimization. 33
mRNA half-life (t1/2)
The mRNA half-life (t1/2) is the time it takes for 50% of a specific mRNA population in a cell to be degraded. It is a critical determinant of the overall amount and duration of protein expression from an mRNA therapeutic or vaccine. 34 A longer half-life provides more opportunities for the mRNA to be translated, thus increasing protein yield. mRNA half-life is influenced by a multitude of factors, including:
Sequence elements within the UTRs (eg, stabilizing elements or destabilizing AREs in the 3′ UTR. 35
The length and integrity of the poly(A) tail 36 (segmented poly(A) tails or circularization).
Codon optimality within the CDS. 18
The overall secondary structure and GC content of the mRNA. 37
The presence of chemical modifications (eg, m1Ψ). 38
Unpaired probability/average unpaired probability
The unpaired probability of a nucleotide refers to the likelihood that this specific nucleotide is in a single-stranded (unpaired) state when considering the ensemble of all possible secondary structures the RNA molecule can adopt. The average unpaired probability (AUP) is the sum of these unpaired probabilities across all nucleotides in the sequence, often normalized by the length of the RNA. 37 The rationale behind using AUP as an optimization metric is that single-stranded regions of RNA are generally more susceptible to hydrolytic cleavage and enzymatic degradation than base-paired regions. Therefore, a lower AUP, indicating a more structured RNA with fewer accessible unpaired regions, is hypothesized to correlate with increased mRNA stability and a longer half-life. 29 Algorithms like RiboTree aim to optimize sequences by reducing AUP, thereby minimizing multi-loop structures and indirectly enhancing mRNA longevity. 37 AUP can be calculated from base-pairing probability matrices, which are standard outputs of many RNA folding prediction packages like ViennaRNA’s RNAfold.
Many of these quantitative metrics serve as valuable, albeit imperfect, proxies for complex in vivo biological processes. MFE, for instance, reflects in vitro thermodynamic stability, while CAI is based on statistical codon preferences. Their predictive power for actual in vivo performance is not absolute. A significant challenge is that optimizing one metric can sometimes negatively affect another; for example, maximizing structural stability (low MFE) through high GC content might create overly rigid structures that impede ribosome movement or cause issues in PCR amplification. 29 This highlights the critical need for multi-objective optimization strategies that explicitly manage these trade-offs. The field is also witnessing an evolution in metrics themselves, with AI and machine learning contributing to the development of more sophisticated, data-driven predictors that aim to capture functional outcomes more directly, such as MRL predicted by models like UTR-LM 33 or overall protein expression predicted by frameworks like RiboDecode. 39 These advanced metrics move beyond simple statistical correlations toward more integrative and functionally relevant assessments of mRNA sequence quality.
We summarize in Table 1 the quantitative metrics discussed above.
Quantitative metrics for evaluating mRNA sequence optimization.

Computational Model
The immense complexity of mRNA sequence design, characterized by a vast search space and multiple interacting biological parameters, necessitates the use of sophisticated computational models. These models aim to predict the performance of mRNA sequences and to guide their optimization toward desired therapeutic outcomes. Over recent years, a diverse array of algorithms and software tools has been developed, ranging from those focused on single objectives like structural stability to advanced AI-driven platforms capable of multi-objective, full-length mRNA optimization.
Tool choice should be driven by the primary optimization goal and the mRNA region under design. If the priority is structural stability/persistence, begin with structure-centric predictors and optimizers and evaluate impact on t1/2. If the goal is maximizing protein output, prioritize methods targeting translation-related readouts: 5′UTR-focused predictors/generators (eg, UTR-LM, UTRGAN, Smart5UTR) guided by MRL/TE, and CDS-focused codon tools (eg, JCat/GeneOptimizer) guided by CAI. When multiple objectives must be balanced (eg, stability vs translation), adopt multi-objective frameworks (eg, LinearDesign, mRNA-LM, integrated tools such as mRNAdesigner) and report trade-offs across MFE/AUP, CAI, MRL, and t1/2. Finally, because in silico scores are imperfect proxies, candidate sequences should be shortlisted by constraints (motifs, GC/structure limits) and then validated experimentally, enabling iterative refinement. All the advantages and limitation have been listed in Supplemental Table 1.
Structure optimization
A primary goal in mRNA design is to enhance its stability, thereby prolonging its intracellular half-life and increasing the potential for protein production. Structure optimization models typically focus on modulating the mRNA’s secondary structure to achieve this.
Protein production optimization
While structure influences stability, the ultimate goal for most mRNA therapeutics is efficient protein production. This category of models focuses on optimizing sequences, primarily the CDS, to maximize the yield of the encoded protein.
Multi-objective optimization
Recognizing that optimal mRNA performance depends on a balance of multiple factors, multi-objective optimization models aim to simultaneously improve several, often conflicting, properties such as stability, translational efficiency, and immunogenicity. This reflects a more mature understanding of mRNA biology, moving away from single-parameter optimization.
UTR optimization
Given the critical role of UTRs in modulating mRNA stability and translation, specialized computational models have been developed to focus specifically on the design and selection of optimal 5′ and/or 3′ UTR sequences. The optimization of the entire mRNA, including both UTRs and the CDS, is increasingly recognized as crucial.
These UTR-focused models often leverage machine-learning techniques, particularly deep learning and generative models, trained on large datasets of known UTR sequences and their associated functional data (eg, MRL from Ribo-seq, expression levels from reporter assays). This allows them to learn complex sequence-function relationships and either predict the performance of given UTRs or generate entirely new UTR sequences with desired characteristics.
The evolution of computational models for mRNA optimization clearly shows a trend from single-objective, often rule-based approaches toward more sophisticated, AI-driven multi-objective strategies. Machine learning, particularly deep learning and large language models (LLMs), is playing an increasingly central role, enabling the capture of complex biological patterns from vast datasets that elude simpler methods. There is also a growing emphasis on optimizing the entire full-length mRNA (5′UTR-CDS-3′UTR) holistically, recognizing the synergistic interactions between these regions. Furthermore, the advent of generative AI models marks a significant shift, empowering researchers to design entirely novel sequences with desired properties, thereby vastly expanding the explorable design space beyond mere selection or modification of existing sequences.
We summarize in Table 2 the algorithms discussed above.
Overview of computational models for mRNA sequence optimization.

Discussion
The rapid ascent of mRNA vaccine technology, particularly highlighted during the COVID-19 pandemic, has underscored the critical importance of rational sequence design. Significant progress has been made in understanding the complex interplay between mRNA sequence elements, their corresponding biological functions, and the ultimate efficacy of the encoded protein, whether it be a vaccine antigen or a therapeutic protein. This journey has seen a shift from relatively simple, single-parameter optimization strategies, such as focusing solely on codon adaptation, to more sophisticated, multi-objective computational approaches. The development and application of AI and machine-learning algorithms have been pivotal in this evolution, providing powerful tools to navigate the astronomically vast sequence space and to begin deciphering the intricate sequence-to-function relationships that govern mRNA stability, translation, and immunogenicity. This review has summarized key biological mechanisms, the quantitative metrics used to guide optimization, and the diverse array of computational models currently employed in this dynamic field.
The journey of mRNA vaccine technology has been transformative, moving from foundational research to global therapeutic impact. This progress has been fueled by an enhanced understanding of RNA biology—from the roles of UTRs and codon usage to the impact of secondary structures and chemically modifications—and by the concurrent development of powerful computational tools. Sequence optimization has matured significantly. Initially, efforts might have focused on individual parameters like maximizing CAI or achieving a target GC content. However, the field now widely recognizes that mRNA performance is a result of many interacting factors. Consequently, the focus has shifted toward multi-objective optimization strategies that aim to simultaneously balance stability, translational efficiency, and immunogenicity. AI and ML have become indispensable in this endeavor, capable of learning from large datasets to identify complex patterns and guide the design of sequences with superior characteristics, a task far beyond the reach of manual or simple algorithmic approaches.
Despite the remarkable advancements, current computational models for mRNA sequence optimization are not without limitations. Bridging the gap between in silico design and in vivo performance remains a significant hurdle.
The field of computational mRNA optimization is rapidly evolving, with several exciting avenues for future development:
Building on these directions, three particularly actionable priorities stand out for the next stage of the field. First, progress will depend on standardized, high-quality experimental datasets with harmonized readouts (eg, matched measurements of stability/half-life, translation outputs such as MRL/TE, and innate immune activation markers), ideally generated under comparable cell types, delivery conditions, and chemical-modification settings, to enable robust model training and benchmarking. Second, future model development should increasingly prioritize explainability (eg, interpretable motif/structure attribution and mechanistic hypothesis generation), so that AI systems can move beyond “black-box” prediction to reveal actionable sequence–function rules. Finally, optimization objectives should be explicitly application-dependent: infectious-disease vaccines may prioritize rapid, high peak antigen expression with controlled innate sensing, whereas cancer vaccines and therapeutic protein replacement may require different trade-offs in durability, dosing frequency, and safety—arguing for tunable, context-aware multi-objective frameworks and benchmarks tailored to each use case.
The “no free lunch” theorem seems applicable to mRNA optimization; there is unlikely to be a single, universally perfect mRNA sequence or a one-size-fits-all optimization algorithm. Different therapeutic applications (eg, a vaccine requiring transient but very high antigen expression versus a protein-replacement therapy needing sustained, moderate expression) will likely demand distinct optimization strategies and involve different trade-offs. This necessitates the development of tunable, context-aware computational design tools. Moreover, the critical feedback loop between computational prediction and experimental validation will continue to evolve. Tighter, more rapid iterations, where high-throughput experimental data continuously refines AI models, will be key to enhancing their predictive accuracy and reliability, transforming AI from a mere design tool into an active partner in the scientific discovery process. Ultimately, the field is moving from “sequence optimization” toward “therapeutic optimization,” where the goal is not just an optimal mRNA molecule in isolation but an optimal therapeutic outcome in patients. This will require future models to integrate a broader range of factors, including delivery system characteristics, route of administration, patient-specific variables, and the underlying biology of the target disease.
Conclusion
The optimization of mRNA sequences stands as a critical frontier in the development of next-generation vaccines and therapeutics. The journey from basic RNA biology to clinically successful mRNA products has been accelerated by the advent of powerful computational tools, particularly those driven by artificial intelligence. These approaches are beginning to unravel the complex “RNA code” that governs an mRNA’s fate and function, enabling the design of molecules with enhanced stability, higher TE, and improved safety profiles. While significant challenges remain in bridging the in silico-in vivo gap and in fully modeling the dynamic nature of RNA regulation, the pace of innovation is rapid. Continued interdisciplinary collaboration among molecular biologists, immunologists, chemists, computational scientists, and clinicians will be paramount. By harnessing the combined power of deep biological understanding and sophisticated computational strategies, the immense potential of mRNA technology to address a wide spectrum of human diseases is poised to be realized, heralding a new era of precision medicine.
Supplemental Material
sj-docx-1-bbi-10.1177_11779322261431255 – Supplemental material for Recent Progress in Sequence Optimization of mRNA Vaccine: Biological Mechanism, Quantitative Metrics, and Computational Model
Supplemental material, sj-docx-1-bbi-10.1177_11779322261431255 for Recent Progress in Sequence Optimization of mRNA Vaccine: Biological Mechanism, Quantitative Metrics, and Computational Model by Yunwei Wang, Yuheng Cai, Zhixing Wu, Jingming Zhang, Lance Turtle and Jia Meng in Bioinformatics and Biology Insights
Footnotes
Ethical Considerations
Not applicable. This article does not report any studies with human participants or animals performed by any of the authors.
Author Contributions
Yunwei Wang: Conceptualization; Investigation; Writing—original draft; Writing—review & editing; Methodology; Visualization; Formal analysis; Data curation; Resources.
Yuheng Cai: Resources; Supervision; Project administration; Writing—review & editing.
Zhixing Wu: Visualization; Writing—review & editing; Supervision.
Jingming Zhang: Writing—review & editing; Supervision.
Lance Turtle: Supervision; Writing—review & editing.
Jia Meng: Conceptualization; Supervision; Resources; Project administration; Writing—review & editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Natural Science Foundation of China [31671373]; Scientific Research Foundation of Nanjing University of Chinese Medicine [013038030001]; XJTLU Key Program Special Fund [KSF-E-51 and KSF-P-02]. This work was supported by the Supercomputing Platform of Xi’an Jiaotong-Liverpool University.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Not applicable. This article is a narrative review and did not generate or analyze new datasets.
AI Usage Declaration
The authors used an AI language model (ChatGPT, OpenAI) to refine and enhance the clarity and readability of the manuscript text. No scientific data were generated, analyzed, or modified using AI tools. The authors take full responsibility for the accuracy, originality, and integrity of the submitted work.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.