
Abstract
Buruli ulcer (BU) is a necrotizing skin disease caused by Mycobacterium ulcerans that produces a virulent lipid toxin, mycolactone, which is detectable in urine. Current diagnostics are time-consuming and require specialized expertise, often leading to delayed diagnosis. This makes it difficult to understand the disease’s spread and plan effective interventions. To facilitate early diagnostic biomarker identification, we used computational methods to identify proteins to the antigen lactone, a product of mycolactone hydrolysis, that could be used to develop rapid diagnostic tests (RDTs). Using AutoDock Vina, we performed a virtual screening of 6 proteins against lactone. Four proteins – N-Acyl homoserine lactonases (4G5X), hyperthermophilic Sulfolobus islandicus PLL SisLac (4G2D), phosphotriesterase (2VC5) and quorum-quenching lactonase (6N9I) – showed strong interactions with lactone, with binding energies ranging from −8.9 to −6.0 kcal/mol. Molecular dynamic simulations used to assess the stability of these protein-lactone complexes showed that natural lactonase and promiscuous phosphotriesterase activities (2VC5) and quorum-quenching lactonase GcL (6N9I) were the most stable. In addition, 2VC5 and 4G5X demonstrated the most flexibility. Overall, the proteins 2VC5, 4G2D and 4G5X showed a strong binding affinity, good stability and favourable interactions with lactone. These findings suggest that these proteins could serve as the basis for developing rapid, noninvasive RDTs for BU disease.
Introduction
Buruli ulcer (BU) is a debilitating necrotizing disease of the skin caused by Mycobacterium ulcerans that produces a lipid toxin mycolactone which is responsible for disease symptoms. 1 It ranks as the third most common mycobacterial infection worldwide after leprosy and tuberculosis. 2 Over 98% of BU cases are reported in at least 33 countries within tropical, subtropical and temperate regions in Africa, South America, Western Pacific regions, Australia and Japan. 3 The annual number of suspected BU cases reported globally was around 5000 cases up until 2010 and has witnessed a general decline to reach 2271 in 2019, 1458 in 2020 and 1370 in 2021. 4 The reductions seen in 2020 and 2021 could be linked to the impact of COVID-19 on active detection activities; however, as there has been an overall decline from 2010, it is believed by some scientists it could also be due to changes in the rainfall patterns in the high endemic West African countries Benin, Ghana and Côte d’Ivoire. 5
The mode of transmission of BU remains elusive, although a strong association with water bodies 6 and the likely involvement of Acanthamoeba species have been reported. 7 Typically, BU manifests initially as a painless nodule that progresses with no pain and fever, to a large painless area of induration or oedema to ulceration within 4 weeks. 8 If left untreated, it expands into a painless, extensive ulcer and, in severe cases, may result in lifelong disabilities and deformities. 9 Current first-line therapy involves a combination of antibiotic rifampicin and clarithromycin treatment 10 and complementary surgery, wound care and prevention of disability are advocated. 11
Differential clinical diagnosis is important in BU treatment, as it may occur with other ulcers, including tropical phagedenic ulcers in which fusiform bacilli and spirochetes predominate in the bacterial flora; chronic lower leg ulcers due to arterial and venous insufficiency (often in elderly populations); diabetic ulcers; cutaneous leishmaniasis; ulcerative yaws; and ulcers caused by Haemophilus ducreyi. 12 Confirmed BU diagnosis relies on 4 methods, namely, IS2404 polymerase chain reaction (PCR), culture, microscopy and histopathology.12,13 However, PCR and histopathology for routine diagnoses require well-equipped facilities and skilled personnel. 14 In rural areas, limited resources can lead to delayed diagnoses, missed diagnoses and late treatment. 15 Moreover, it is estimated that BU has an average incubation period of 4.8 months (range = 2-9 months) 16 and this knowledge is important for understanding the epidemiology of BU and the planning of its interventions. Early detection is also vital because BU primarily affects children.
An epidemiological study would ideally require rapid, noninvasive and cost-effective diagnostic techniques that can be implemented en-masse and in resource-limited settings. The World Health Organization’s Diagnostic Technical Advisory Group in 2019 recommended rapid point-of-care tests with mycolactone, bacterial protein or DNA as the target analytes for individual diagnosis at the primary healthcare/community level. 17 However, mycolactone is unstable, and hence, it will be difficult to capture for diagnosis outside of the human body. 13 Also, mycolactone is known to have a very short life in humans, which limits detection in its entirety in the blood. To overcome these challenges, we hypothesize that mycolactone is hydrolysed to the core lactone ring and 2 polyketide chains in the blood. 18 Incidentally, the core lactone exhibits cytopathic, cytotoxicity and immunosuppression as observed in BU.19,20
The core lactone, a 12-membered cyclic ring (C1-C11) within mycolactone, is a critical structural component formed during the toxin’s biosynthesis via a 2-polyketide-synthase model. Unlike the full mycolactone molecule, which includes 2 polyketide side chains, the core lactone exhibits independent cytotoxic properties, making it a biologically significant entity. 20 The core lactone induces cytopathic effects on murine fibroblasts, including cell rounding, cell cycle arrest and apoptosis, at concentrations around 12 500 ng/mL, although it is less potent than mycolactone A/B (0.01 ng/mL). 19 The core lactone’s presence in M ulcerans isolates from regions like Africa and Malaysia correlates with variations in disease severity, such as more invasive forms leading to osteomyelitis. 19 While mycolactone is responsible for tissue necrosis, immunosuppression and painlessness in BU lesions, the core lactone’s bioactivity suggests it could contribute to these effects if present in vivo, either as a biosynthetic precursor or as a degradation product. 21 Studies indicate that mycolactone fragments into the core lactone during analytical processes like mass spectrometry, suggesting greater stability compared with intact mycolactone. 22 This stability, combined with its bioactivity, positions the core lactone as a promising target analyte for rapid diagnostic tests (RDTs) in resource-limited settings. 17
The development of a noninvasive diagnostic method for BU would be a multistage process encompassing biomarker discovery, in vitro assay validation, ultimate design and development and testing/evaluation. Computational approaches offer a powerful toolset to shorten the process of biomarker discovery. Molecular docking allows for the prediction and examination of molecular interactions, particularly the binding of small molecules to larger macromolecules. 23 Computational approaches have previously been used in biomarker discovery, where molecular modelling augmented experiments towards the discovery of a biomarker for the diagnosis of sepsis. 24 The authors performed molecular docking of aptamers and procalcitonin (PCT) to provide detailed insights into the binding mechanism of PCT, which was characterized by the formation of strong hydrogen bonds and hydrophobic interactions with selected aptamers. Herein, we used molecular methods to identify proteins that bind to lactone as the target analyte for the development of an RDT for BU.
Materials and Methods
Lactone structure retrieval
The canonical Simplified Molecular Input Line Entry System (SMILES) of mycolactone was retrieved from Puchem (ID 5282079), a database that contains information on chemicals against their biological assays. Its 2-dimensional (2D) structure was generated using DrugBank by removing the 2 fatty acyl chains to retain the core lactone ring. 25 The modified structure was converted to a 3-dimensional (3D) conformation and energy-minimized using the MMFF94 force field implemented in Open Babel (version 3.0.0) on a Linux system to obtain the lowest energy conformation. The modified structure was saved in .pdb format and visualized using PyMOL (PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC).26,27
Protein structure retrieval and preparations
A structured literature search was conducted on Google Scholar using combinations of keywords such as ‘lactonase binding to lactone’, ‘hydrolase lactone interaction’, ‘quorum-quenching lactonase’ and ‘lactone hydrolysis enzymes’, provided that lactonases have the ability to bind unto lactone through the ester bond in the lactone ring.28,29 The search was refined to include studies that reported crystal structures of enzymes with experimentally verified activity towards lactone or lactone-containing substrates. Proteins were prioritized based on functional relevance, structural quality, catalytic diversity and previous biological association. Preference was given to lactonases and hydrolases whose primary function involves catalysing the hydrolysis of lactones or similar cyclic esters. Only proteins with experimentally determined 3D structures available in PDB and resolutions of 3.0 Å or better were included. To capture structural diversity among lactone-binding enzymes, representatives from different enzyme classes, including metallo-lactonases, phosphotriesterases and α/β-hydrolases, were selected. Munc18-b (4CCA) was included based on previous reports of its binding to mycolactone, serving as a reference molecule. 30 Following these criteria, 6 proteins were selected: 4G5X, 4G2D, 2VC5, 7RIS, 6N9I and 4CCA. Among these, 6N9I, 4G5X and 2VC5 were chosen for their high catalytic activity as natural lactonases, while 7RIS and 4G2D represented structurally distinct lactone-interacting enzymes. Several other lactonases with potential relevance were identified during the search; however, they were excluded because of incomplete structural information, unresolved catalytic sites or redundancy in function with already selected enzymes. The 6 proteins, as shown in Table 1, were retrieved from the Research Collaboratory for Structural Bioinformatics Protein Data Bank.27,31 The structures of the proteins were viewed in PyMOL before molecular docking using AutoDock Vina implemented through PyRx (version 0.8). During preparation, water molecules and co-crystallized ligands were removed, and only chain B of the 4 identical chains of the 2VC5 structure was used for the docking and all water molecules and co-crystallized molecules were removed. Chain B of the 2 identical chains of 4G5X was also used for docking, and all water molecules were removed. Chain C of the 3 identical chains in 6N9I was used for docking as well, and all water and co-crystallized molecules were removed. All water and co-crystallized molecules in 4G2D, 7RIS and munc18b were also removed before docking. As the chains had identical amino acid sequences, the presence of ligands and binding sites, different chain identifiers were used for the docking.
List of the 6 proteins used for molecular docking.

The table shows their PDB IDs, names, methods used to determine their structures and the resolutions obtained.
Validation of docking protocol
The docking protocol was validated by using the 6N9I and 2VC5 complexes. The GOL ligand of the co-crystallized 6N9I structure was removed and redocked in AutoDock Vina. The co-crystallized complex and redocked complex were aligned using the BIOVIA Discovery Studio 25.1.0, and the root mean square deviation (RMSD) of the 2 ligands was then calculated. The redocked complex was again superimposed onto the co-crystallized complex using LigPlot + v.2.2 without any adjustments to identify the common residues the ligand interacted with in both complexes. The superimposed 2D structure is shown in the superimposed amino acids of the complexes encircled in red. This approach accessed AutoDock Vina’s ability to predict the binding pose.
Molecular docking
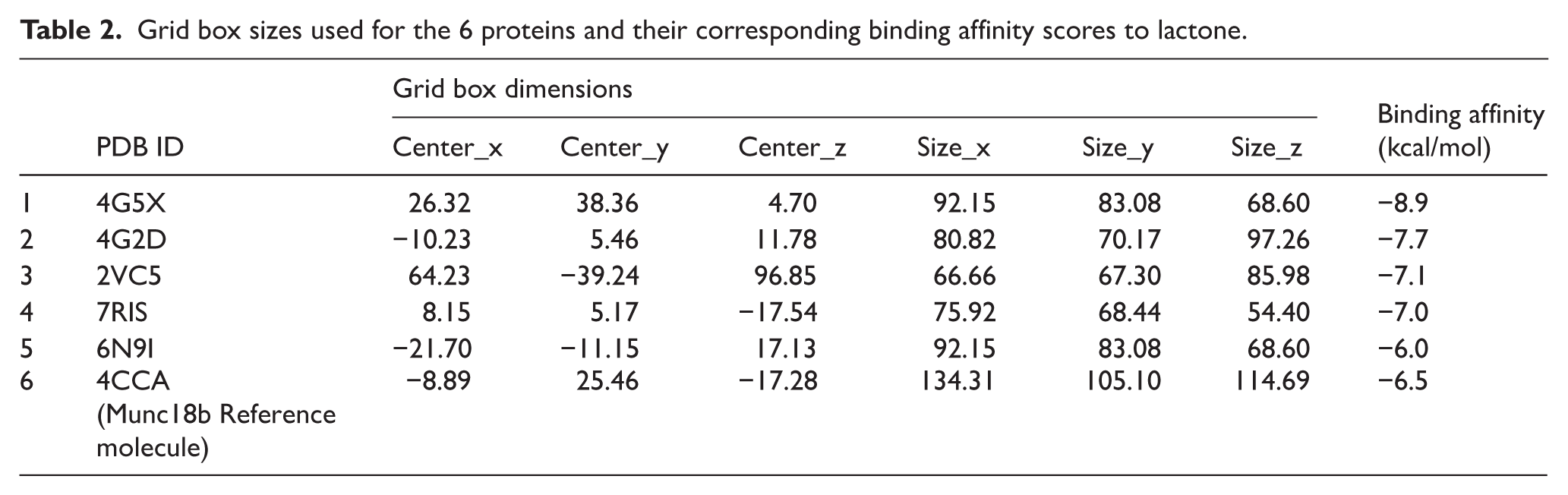
AutoDock Vina32,33 interfaced with PyRx version 0.8 34 was used to run the molecular docking and the poses were visually assessed in PyMOL (PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC. 35 The target was converted to AutoDock Vina files using the ‘make macromolecule’ option. The compounds to be screened against the targets were subjected to energy minimization using the universal force field and conjugate gradient algorithm in 200 steps and were converted to Protein Data Bank partial charge and atom type (.pdbqt) file format using Open Babel. 36 For each protein, a specified grid box encompassing the active site of each protein was created with respective pocket dimensions as shown in Table 2. The default exhaustiveness of 8 for AutoDock Vina computations was used as the limit of docking conformations (Figure 1).
Grid box sizes used for the 6 proteins and their corresponding binding affinity scores to lactone.

Methodology schema for the study detailing the key steps used to screen compounds against the targets.
Characterization of the mechanism of binding
LigPlot+v1.4.5 was used to characterize the interactions between the protein and the selected hits by generating 2D schematic diagrams of protein-ligand interactions of the complexes generated from PyMOL.26,37 The hydrogen bonds were depicted as green dashed lines and the arcs with spokes radiating towards the ligands represented hydrophobic interactions.
Molecular dynamics simulation of protein-ligand complexes
Molecular dynamics was performed using the GPU version of AMBER 18 with an incorporated PMEMD module38,39 following standard protocols as elaborated in these computational studies.40-42 All 6 complexes generated from molecular docking were prepared from molecular simulation. The corresponding unbound (apo) forms of the proteins were also prepared for simulation.
Parameterization of lactone was carried out using the ANTECHAMBER module wherein atomic partial charges (AM1BCC) Gaff using the bcc charge scheme were added.43,44 The protein structures were also parameterized by the FF14SB AMBER force field. 45 Using the LEAP module, hydrogen atoms were also added, while the entire system was neutralized by adding counter ions (Na+, Cl–), followed by a subsequent generation of ligand, protein and complex topologies as well as parameter files for each of the molecules. The systems were precisely solved with water molecules using the TIP3P orthorhombic box size of 8Å.46,47 Solvated complexes were minimized initially for 2000 minimization steps applying a restraint potential of 500 kcal/mol and then fully minimized for another 1000 steps of steepest descent without restraint. This was followed by the gradual heating of the systems from 0 to 300 K for 50 ps, after which they were equilibrated for 500 ps while the temperature and pressure were kept constant at 300 K and 1 bar, respectively.48,49 This was followed by MD production runs of 300 ns for each system, during which the SHAKE algorithm50,51 was used to construct all atomic hydrogen bonds. The MD simulation was initiated using a time step of 1 fs and coordinates saved at 1 ps interval, followed by subsequent analysis of trajectories using the integrated PTRJ and CPPTRAJ module.52,53 Visualization of the complexes and data plots was carried out using the graphical user interface of UCSF Chimaera54,55 and Microcal Origin Analytical software. 56
Post-molecular dynamic simulation analysis
After a 300 ns MD simulation run, coordinates of all the simulated systems were saved at 1 ps intervals, followed by subsequent analysis of trajectories using the integrated PTRJ and CPPTRAJ module.53,57 Root mean square deviation, root mean square fluctuation (RMSF) and thermodynamic calculations were calculated and plotted for each of the resultant trajectories. Visualization of the complexes and data plots was carried out using the graphical user interface of UCSF Chimera, Discovery Studio 32 and Microcal Origin analytical software. 58
Thermodynamics calculations
The molecular mechanics/generalized born surface area (MM/GBSA) method was used to calculate binding free energy.59,60 The binding free energy (ΔGbind) of lactone in each complex was calculated as follows:


where ΔGbind is the summation of the gas phase and solvation energy terms less the entropy (TΔS) term.

ΔEgas is the sum of the AMBER force field internal energy terms ΔEint (bond, angle and torsion), the covalent van der Waals (ΔEvdw) and the non-bonded electrostatic energy component (ΔEelec). The solvation energy is calculated from the following equation:


The polar solvation contribution is represented as GGB, and Gnon-polar is the non-polar solvation contribution and is calculated from the solvent accessible surface area (SASA), obtained using a 1.4 A° water probe radius. The surface tension constant (c) was set to 0.0072 kcal/mol and b to 0 kcal/mol. Per-residue decomposition analyses were also carried out to estimate the individual energy contribution of residues of the binding pocket towards the affinity and stabilization of lactone.
Results and Discussion
Structure of mycolactone
The canonical SMILES of mycolactone was obtained from PubChem CID 5282079, which has a molecular weight of 743 g/mol. The chemical structure of mycolactone A/B has a core cyclic lactone ring (C1-C11) and 2 polyketide-derived highly unsaturated acyl side chains. The upper ‘Northern’ chain consists of C12-C20 and the longer ‘Southern’ chain is numbered C1′-C16′ (Figure 2). The SMILES from PubChem was input into the DrugBank to obtain the structure of mycolactone. The structure of lactone was obtained by eliminating the 2 chains of fatty acids as expected during hydrolysis. The carbon ring lactone was saved as a pdb file and used for molecular docking.

The chemical structure of mycolactone A/B shows the core lactone ring and polyketide side chains (Sarfo et al 19 ).
Docking protocol validation
The ligands of the co-crystallized structure and the redocked structure were aligned in BIOVIA Discovery Studio 25.1.0, and RMSDs of 0.964 and 0.835 Å for 6N9I and 2VC5, respectively, were obtained, as shown in Figure 3, which signifies AutoDock Vina’s ability to predict the pose of a ligand in a binding pocket, as an RMSD less than or equal to 2.0 Å is a good threshold (Adams et al 57 ). The Ligplot in Figure 4 demonstrates the residues the co-crystallized complex and redocked complex have in common after they have been superimposed. For 6N9I, 4 residues were common to both complexes: 3 hydrophobic interactions with Ala99, Tyr239 and Trp200, and Trp158 formed a hydrophobic bond with the co-crystallized complex. Similarly, 4 residues, Phe229, Asp202, Asn172 and Ala173, were common in both the crystallized and redocked structure of the 2VC5 protein.

Superimposition of the co-crystallized complex (yellow) with the redocked complex as viewed in Discovery Studio. (A) redocked 2VC5 (green); (B) redocked 6N91 (blue). The close alignment between the 2 complexes indicates good docking accuracy, suggesting that the docking protocol reliably reproduces the experimental binding pose.

Superimposed Ligplots comparing the interactions between the co-crystallized complex and the redocked complex with UnK1(N) as GOL. (A) 2VC5; (B) 6N91. Residues circled in red represented the predicted molecular interaction of the co-crystallized and redocked complex.
Molecular docking
A literature review of proteins that bind to mycolactone 30 and lactone generated 6 proteins with PDB IDs: 4CCA, 7RIS, 2VC5, 6N9I, 4G5X and 4G2D. The binding mode of each protein with an energy-minimized lactone was predicted using AutoDock Vina. For each protein, a specified grid box encompassing the active site of each protein was created with respective pocket dimensions as shown in Table 2. Using AutoDock Vina, the binding modes of lactone bound to each of the proteins were predicted with respective docking scores ranging from −6.0 to −8.9 kcal/mol as shown in Table 2. A binding score of −6.0 kcal/mol was considered favourable enough to discriminate between specific and nonspecific protein-ligand interactions. 61 The docking scores corroborate the sum of the intermolecular forces that characterize ligand-protein interactions. The lower the docking score, the stronger the binding affinity of the ligand towards the protein. 30 Lactone was predicted to assume favourable binding modes towards all 6 proteins, suggesting stronger binding affinities. Previous computational exploration of Munc18b determined that mycolactone was a strong binder with a docking score of −8.5kcal/mol 30 ; hence, it was used in this study as a standard for validating the docking process.
Protein-ligand interactions of lactone complexes with the identified proteins
The prominent interactions that were formed between lactone and the 6 proteins, as generated by LigPlot before molecular dynamics simulations, were hydrogen bonds and hydrophobic bonds, as shown in Table 3 and Figure 5. Lactone interacted with 2VC5 at the residue Try97 forming 1 hydrogen and 9 hydrophobic bonds, 4G2D interacted at the Try99 residues with 1 hydrogen bond and 8 hydrophobic bonds and 4G5X interacted at the Leu103 residue with 1 hydrogen bond and 6 hydrophobic bonds, with 6N9I at the His120 and Tyr158 forming 2 hydrogen bonds and 1 hydrophobic bond, and with 7RIS at Arg141 forming 2 hydrogen bonds of lengths 3.05 and 3.17 with the same residue and 7 hydrophobic bonds, and interacted with munc18b at Thr107 forming 1 hydrogen bond and 8 hydrophobic bonds.
The intermolecular bonds interaction a between lactone and the 6 proteins.

All residues have their chain identifiers in brackets to indicate which of the identical chains was used for docking.

Two-dimensional LigPlot representation showing the interactions between lactone (Unk0, shown in purple) and each of the 6 proteins: (A) 7RIS; (B) 6N9I; (C) 4G5X; (D) 2VC5; (E) 4G2D; and (F) Munc18B. Hydrogen bonds are indicated by green dashed lines, while hydrophobic contacts are represented by red arcs with spokes. Residues involved in the interactions are labelled with their respective amino acid codes and chain identifiers (in brackets). The strong hydrogen bonding interactions highlight key residues within the binding pockets that stabilize lactone binding, suggesting possible regions of functional importance.
Previous studies have reported that lactonase enzymes exhibit binding affinity towards various lactone-containing molecules through interactions involving the ester carbonyl oxygen and key catalytic residues such as His, Asp and Glu. 62 Similar binding mechanisms were observed in this study, where hydrogen bonding and hydrophobic interactions stabilized the lactone-protein complex. Comparable ester-hydrogen bond interactions between lactone carbonyl oxygens and catalytic residues have been reported for quorum-quenching lactonases: crystal structures and mechanistic studies of AiiA and related enzymes show the lactone carbonyl acting as a hydrogen bond acceptor to active site residues (Tyr, His, Asp/Glu), and recent biochemical/docking work demonstrates that bacterial lactonases can accommodate and hydrolyze γ-butyrolactone-type substrates via similar H-bonding and hydrophobic contacts.63-65 Also, Dor et al 29 highlighted the versatility of lactonases in recognizing diverse lactone substrates through flexible active sites, which may explain the binding observed between lactone and the selected proteins in this study. Furthermore, the binding of mycolactone’s lactone ring to Munc18-b, as also reported by Kwofie et al, 30 further validates the biological plausibility of the docking results. Collectively, these findings indicate that the interactions predicted in this study align closely with established patterns of lactone recognition by lactonase and related proteins.
Molecular dynamics and thermodynamics calculations
Conformational changes of potential biomarker proteins upon lactone binding
Structural changes that occurred upon the binding of lactone to the proteins were assessed using parameters such as the C-α RMSD and C-α RMSF, which are suitable to evaluate the stability, motions and structural displacement of constituent residues. As shown in Figures 6 and 7, and Table 4, a comparison of the unbound conformation of each protein with their respective lactone-bound forms showed that the 4G2D, 4G5X, 7RIS and munc18b complexes showed relatively higher RMSD of the complexes, suggesting that the binding of lactone possibly increased the atomistic deviation of the protein hence decreasing their stability. On the contrary, the lactone-bound complexes with 2VC5 and 6N9I showed a decrease in RMSD over the simulation period, suggesting the binding implications of lactone on these proteins could include impeding atomistic deviations and thus inducing some form of structural stability.

RMSD and RMSF plots show the dynamic behaviour of the unbound proteins and lactone-protein complexes (2VC5, 4G2D and 4G5X) over 300 ns molecular dynamics simulations. The RMSD graphs illustrate the comparative structural stability and motion of the unbound and bound forms, while the RMSF graphs depict residue-level flexibility variations upon ligand binding.

RMSD and RMSF plots show the dynamic behaviour of the unbound proteins and lactone-protein complexes (6N9I, 7RIS and Munc18B) over 300 ns molecular dynamics simulations. The RMSD graphs illustrate the comparative structural stability and motion of the unbound and bound forms, while the RMSF graphs depict residue-level flexibility changes upon ligand binding.
Average RMSD and RMSF scores of simulated systems over the 300 ns simulation.

Likewise, the flexibility of individual amino acid residues of the proteins over the simulation period was assessed over the simulation period by calculating the RMSFs of each amino acid as presented in Table 4 and Figure 6. The lactone-bound complexes in 2VC5, 4G5X, 7RIS and munc18b were relatively higher compared with their respective unbound proteins, suggesting the binding of lactone to these proteins increases the flexibility of residues in these proteins. On the contrary, the lactone-bound complexes with 4G2D and 6N9I showed a decrease in average RMSF relative to their respective unbound conformations, suggesting the binding of lactone could be characterized by impeding residue motions. Decreased residue fluctuations are consistent with structural alterations that could interrupt or interfere with crucial protein functions. In all, it could be inferred that the binding of lactone to the selected proteins induces varying structural implications from one target to the other. Overall, 4G5X, 2VC5 and 4G2D have stronger van der Waal bonds and good stability and flexibility. These proteins when biotinylated can be used in RDT instead of an antibody to detect the presence of the lactone biomarker by giving off a colour, as in the works of Diaz-Perlas et al. 66
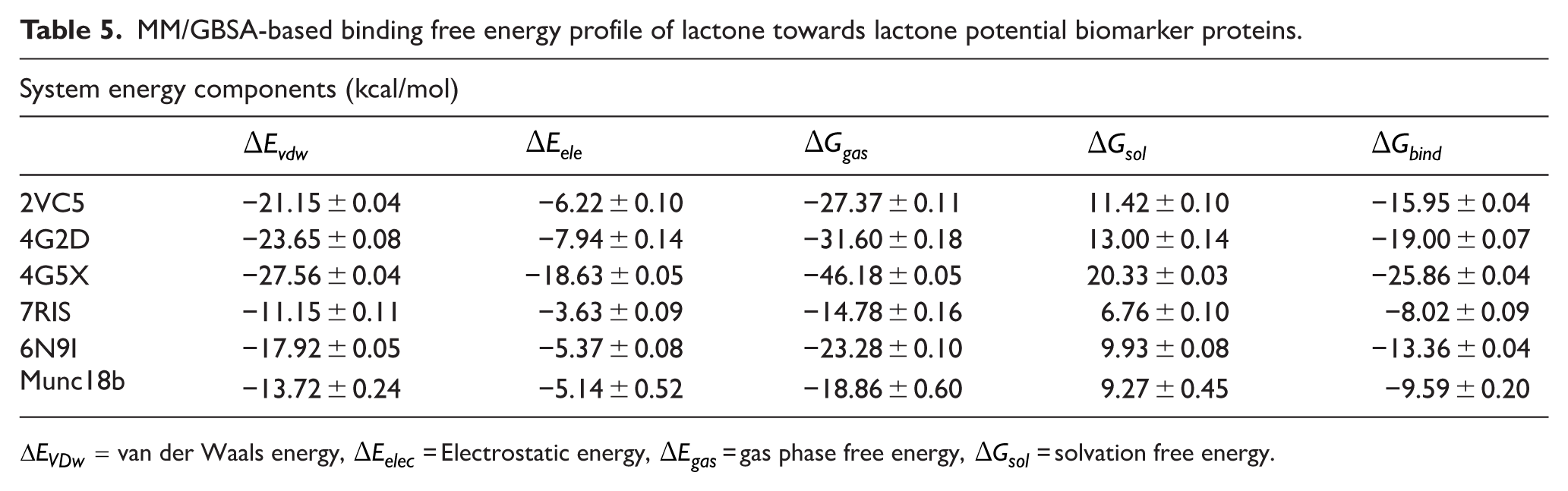
Lactone shows strong binding free energies towards potential biomarker proteins
We calculated the binding free energies of lactone to ascertain its affinity and corresponding stability in complex with the respective proteins. Using the MM/GBSA method, the binding free energies of lactone were −15.9, −19, −25.86, −8.02, −13.36 and −9.59 kcal/mol to 2VC5, 4G2D, 4G5X, 7RIS, 6N9I and munc18b, respectively (Table 5). Among the complexes, lactone was shown to bind stronger to 4G5X characterized by relatively stronger van der Waals (−27.56 kcal/mol) and electrostatic interactions (−18.63 kcal/mol), whereas the lowest binding affinity was observed in the interaction with 7RIS. The strong binding affinity calculated is also consistent with the strong intermolecular interactions observed in the LigPlot analysis.
MM/GBSA-based binding free energy profile of lactone towards lactone potential biomarker proteins.
Per-residue energy decomposition and binding dynamics reveal crucial residues in the binding of lactone to the proteins
Per-residue energy contribution to total binding was also calculated over the simulation period to highlight crucial residues in the binding of lactone to the potential biomarker proteins. The stability of the proteins to their respective targets is influenced by the various interactions and energy contributions of the crucial residues within each binding site. Using the MM/GBSA approach, individual residue contributions with much emphasis on electrostatic van der Waals (vdW) and their overall total energies contributed to the binding of lactone. Residues with interaction energies below −0.8 kcal/mol are generally considered crucial for molecular recognition, high affinity binding and stability of small molecular compounds. Within the 2VC5 complex, lactone engaged in π-alkyl interaction with Ala266 and Leu228 (Figure 8A). The π-alkyl interaction enhances the binding affinity of bound ligands by providing additional noncovalent forces that complement other interactions, suggesting they could have accounted for the strong binding free energy of lactone towards 2VC5. Similarly, in the 4G2D complex, lactone maintains π-alkyl interactions with Ala266 and Leu228 in addition to a π-π stacked interaction with Trp278 9F (Figure 8B). The additional π-π stacked interaction with Trp278 could have accounted for the relatively higher binding of lactone to 4G2D than the 2VC5 complex.

Per-residue decomposition plots showing individual energy contributions towards the binding of lactone and an average snapshot from 250 ns MD simulation showing intermolecular interactions between lactone in 2VC5 (A) and 4G2D (B) complex, respectively.
In the 4G5X complex, the stronger binding of lactone relative to the other complexes was characterized by the formation of π-alkyl interaction with Phe221, Met144 and Ala147 (Figure 9A). This was in addition to a conventional hydrogen bond interaction with Asn33 and π-π interaction with Phe221. The binding of lactone in the 6N9I complex, on the contrary, involved π-π interaction with Glu141, Ala157 and Tyr158 (Figure 9B). In addition, a carbon-hydrogen bond was formed with Ala157. These contributed to the formation of electrostatic interactions but did not influence the overall total binding as shown and corroborated by the weak total binding relative observed with the other systems.

Per-residue decomposition plots showing individual energy contributions towards the binding of lactone and an average snapshot from 250 ns MD simulation showing intermolecular interactions between lactone in 4G5X (A) and 6N9I (B), respectively.
The binding of lactone in the 7RIS complex was mediated by pi-alkyl interactions with Pro189 and π-π interactions with Arg141 (Figure 10A). This was in addition to salt bridges with Arg141 and a conventional hydrogen bond with Lys140. In the munc18b complex, the binding was also characterized by carbon-hydrogen interactions with Thr99 and Ala149 together with a salt bridge with Leu135 (Figure 10B).
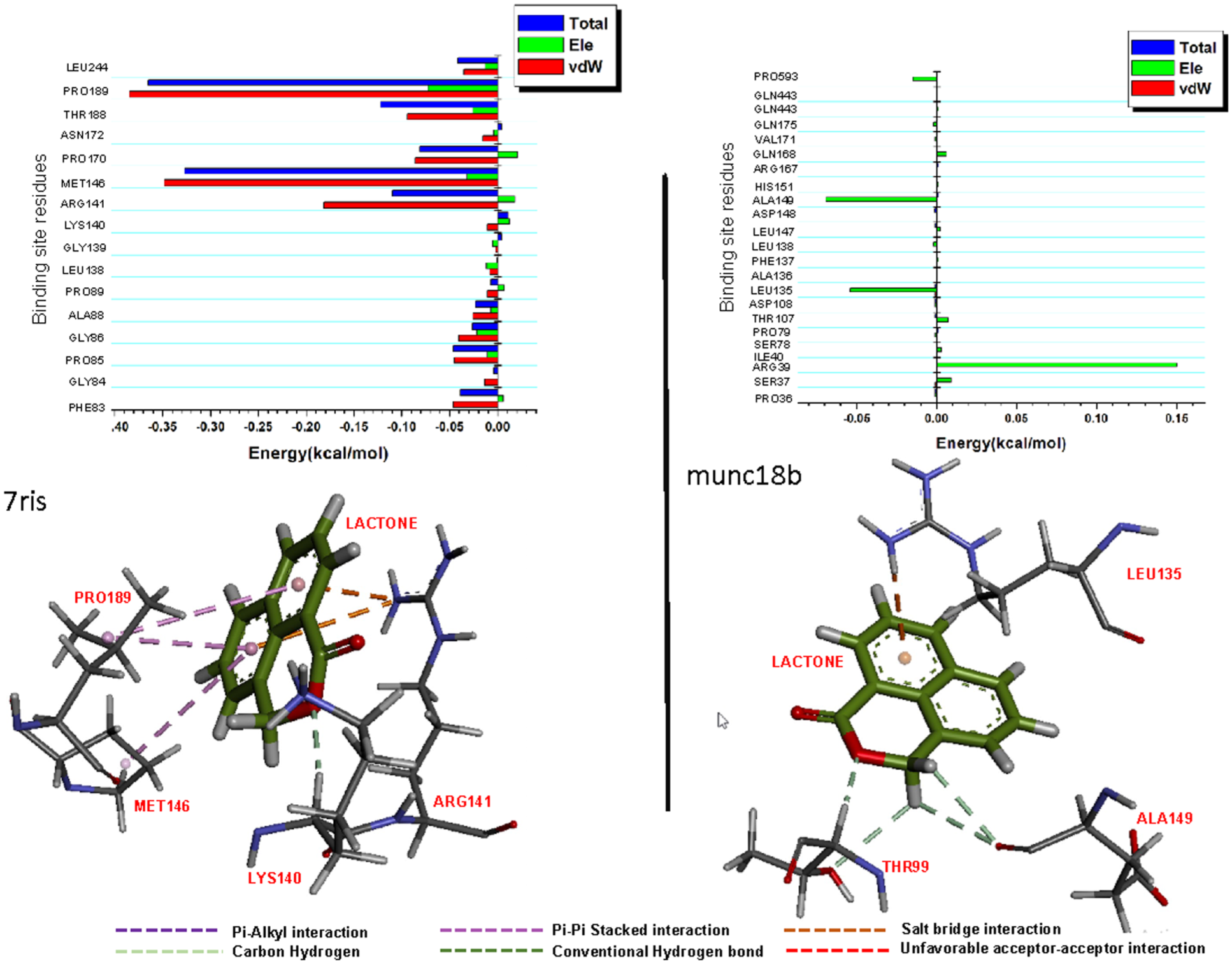
Per-residue decomposition plots showing individual energy contributions towards the binding of lactone and an average snapshot from 250 ns MD simulation showing intermolecular interactions between lactone and 7RIS (A) and munc18b (B), respectively.
Limitations of the Study
The computational methods used in this study offer valuable insights into the potential development of a noninvasive diagnostic approach for BU disease; the results remain preliminary and require experimental validation before diagnostic application. Computational docking and MM/GBSA scores provide theoretical estimates of binding affinity; however, low binding energy alone does not guarantee specificity or detectability in biological fluids. Such results may include false positives, that is, complexes that appear stable in silico but fail to exhibit meaningful interactions under physiological conditions. Therefore, these computational predictions should be interpreted with caution and regarded as hypotheses to be tested experimentally.
To experimentally verify these computational findings, both in vitro and in vivo assays are proposed. An enzyme-linked immunosorbent assay could be used to quantify lactone-protein interactions with high sensitivity. In this setup, microtiter plate wells would be immobilized with lactone, and proteins conjugated with an enzyme such as horseradish peroxidase would be introduced to assess specific binding. The resulting colorimetric or fluorescent signal could be used to construct a standard curve of fluorescence intensity versus lactone concentration, allowing determination of binding affinity in unknown samples.
Complementary in vitro assays such as surface plasmon resonance or isothermal titration calorimetry could further characterize the kinetics and thermodynamics of these interactions, providing an additional layer of validation. For in vivo confirmation, animal model studies will be necessary to evaluate lactone’s biodistribution, stability, and detectability in physiological environments, ensuring its reliability as a diagnostic probe under real biological conditions.
Conclusion
This study represents a significant step forward in the pursuit of rapid and efficient diagnostic tools for BU, a debilitating infectious disease caused by M ulcerans. With a focus on the in silico identification of potential proteins that bind to lactone, this research offers promising insights into proteins that can be used for the development of point-of-care RDT kits for this neglected tropical disease. Using molecular modelling methods, potential biomarker proteins of BU with PDB codes 2VC5, 4G5X and 4G2D were identified. The binding of the identified proteins was characterized by strong intermolecular interactions with lactone corroborated by high binding free energies estimated over a 300 ns simulation period. The development of RDTs based on these compounds could provide rapid, noninvasive and cost-effective methods for early detection of BU, particularly in resource-limited settings where access to traditional diagnostic facilities is limited. However, further in vitro experiments are recommended to attest their potential as biomarker-binding proteins for BU diagnosis.
Footnotes
Acknowledgements
The authors are appreciative to the West African Centre for Cell Biology of Infectious Pathogens, University of Ghana, for permitting the use of Zuputo, their high-performance computer equipment. This work is dedicated to the memory of Professor Wilson, who died during the first revision of this paper, and the authors are grateful for his mentorship and guidance of numerous students and researchers.
Ethical Consideration
The study did not involve any animals or humans.
Author Contributions
Erica A Akanko: Methodology; Data curation; Writing – original draft; Visualization.
Clement Agoni: Methodology; Writing – original draft; Validation.
George Hanson: Writing – review & editing.
Henrietta Esi Mensah-Brown: Supervision.
Kwabena Kan-Dapaah: Supervision.
Claude Fiifi Hayford: Supervision.
Cletus Fiifi Adams: Supervision.
Lydia Mosi: Conceptualization; Writing – review & editing; Resources; Supervision.
Samuel K Kwofie: Supervision; Resources; Conceptualization.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Data Availability Statement
All data regarding this research are included in the paper.