
Abstract
An advanced graph neural network (GNN) is of great promise to facilitate predicting Poly ADPribose polymerase inhibitors (PARPi). Recent studies design models by leveraging graph representations and molecular descriptor representations, unfortunately, still face challenges in comprehensively capturing spatial relationships and contextual information between atoms. Moreover, combining molecular descriptors with graph representations may introduce information redundancy or lead to the loss of intrinsic molecular structures. To this end, we proposed a novel Residual Reconstruction Enhanced Graph Isomorphism Network (R2eGIN) learning model. Specifically, we first designed a residual GIN to learn molecular representations, reduced the impact of vanishing gradients, and enabled the model to capture long-range dependencies. Then, the reconstruction block, by predicting adjacency matrices and node features, was adopted to reconstruct the input graph. To prove the effectiveness of the proposed model, extensive experiments were conducted on 4 data sets of PARPi and compared with 7 existing models. Our evaluation of R2eGIN, conducted using 4 PARPi data sets, shows that the proposed model is comparable to or even outperforms other state-of-the-art models for PARPi prediction. Furthermore, R2eGIN can revolutionize the drug repurposing process through a substantial reduction in the time and costs commonly encountered in traditional drug development methods.
Keywords
Introduction
Recent discoveries regarding Deoxyribonucleic Acid (DNA) damage repair processes have led to increased attention from researchers and medicinal chemists toward novel antitumor PARPi.1-4 Poly(ADP-ribose) polymerase (PARP), a protein superfamily comprising 17 enzymes, plays a crucial role in DNA damage repair and genome stability maintenance, as well as regulating various cellular processes, including DNA damage signaling, chromatin remodeling, transcription, replication fork stabilization, detection of unligated Okazaki fragments during replication, inflammation, and metabolism.5,6 PARP-1 has garnered attention in the therapeutic field due to its overexpression observed in various cancers, including ovarian, breast, skin, colorectal, lung, and others. 7 PARP-1 plays a key role in cell differentiation, gene transcription, inflammation, mitosis, and cell death, contributing to the antitumor activity of PARP-1 inhibitors. 8 The interaction between PARPi and the β-NAD + binding site on PARP1 results in the inhibition of the enzyme’s function. Consequently, DNA repair mechanisms are disrupted, leading to the accumulation of genetic errors and ultimately cell death.9,10 PARP-2 plays a significant role in regulating epigenetic modifications, promoting cell proliferation, and mediating inflammatory processes. According to existing studies, PARP-2 inhibition is strongly associated with hematological toxicity. 1 In addition to PARP-1 and PARP-2, whose roles are associated with DNA damage repair, there are also PARP-5a (TNKS1) and PARP-5b (TNKS2), which are members of the Tankyrase (TNKS) family. PARP-5b is recognized as a key factor involved in maintaining genetic stability, as it controls DNA damage repair and cell cycle regulation. More specifically, PARP-5B regulates various mitotic functions, including centrosome function, mitotic spindle assembly, mitotic checkpoints, telomere length, and telomere cohesion. 6 The important role of PARP-5a and PARP-5b also includes inhibiting their DNA-binding capacity and inducing Wnt/β-catenin signaling pathway regulation through PARylation-mediated proteasomal degradation. 11 Most importantly, a growing body of research indicates that PARP inhibitors have significant potential in treating noncancer conditions, such as ischemic stroke, 12 cardiovascular disease, 13 and diabetic retinopathy. 14
Recent research highlights the significant potential of PARP inhibitors, such as CVL218, which is undergoing phase I clinical trials, in inhibiting SARS-CoV-2 replication 15 and combating the harmful effects of COVID-19 through various mechanisms.15,16 Nevertheless, approved PARP inhibitors face challenges in clinical use, including toxicity, selectivity issues, and drug resistance.17-19 Therefore, the development of novel PARP inhibitors for the treatment of both tumor and nontumor diseases is highly necessary.
Traditional drug discovery often necessitates numerous experiments that are time-consuming and costly. Accordingly, computational approaches serve as a viable option for early-stage drug discovery, aiming to reduce wet-lab experiments and minimize costs. In the field of bioinformatics and molecular properties, GNN demonstrate their success and effectiveness.20,21 GNN are capable of exploring intrinsic information embedded within nodes, as well as inter-node relationships, and transforming them into valuable features for modeling. This is particularly suitable for data such as molecules, where molecular structures and functional relationships between molecules correlate with significant biological information.
Although the development of GNN models in molecular property prediction research, including PARP inhibitor prediction, has attracted research attention in recent years, there remains a need to enhance information propagation between layers, enabling models to capture deeper molecular features by leveraging key inter-atomic relationships for PARP inhibitor molecule prediction. Furthermore, as GNN become more complex, models face the vanishing gradient problem due to excessive layer depth. We argue that implementing residual learning and a reconstruction network can improve the model’s capacity to predict PARP inhibitor molecules. Applying residual learning to the GNN backbone helps address the vanishing gradient problem and enhances information propagation between layers, allowing models to capture deeper molecular features. Meanwhile, a reconstruction network enables models to refine molecular representations by preserving spatial structures and topological relationships, thereby improving generalization and molecular property prediction, including in PARP inhibitor identification.
In this study, we introduce the R2eGIN model for the prediction of PARPi. The proposed model is an extension of the graph isomorphism network (GIN), incorporating residual connections and reconstruction blocks to improve PARPi prediction accuracy. This study represents the first application of the GIN approach for PARPi prediction, where the integration of residual connections and reconstruction mechanisms enhances the model’s ability to capture molecular structural relationships and improves the stability of information propagation within the network, thereby enabling more accurate predictions. An ablation study was conducted to analyze the impact of each additional component on prediction performance. Finally, the effectiveness of the proposed model was validated by comparing it with existing methods on the same data sets.
Methods
R2eGIN model
The overview of the R2eGIN model is illustrated in Figure 1, which comprises 3 main parts: a GNN backbone, the residual connection, and a reconstruction block. Each of the parts is discussed comprehensively in the following subsections, respectively.

Overview of R2eGIN.
Graph neural networks
In this study, a GNN-based approach was employed to aggregate nodes and edges, thereby generating graph-level representations. The GNN model employed in this paper was an extended version of the GIN. We considered a set of molecules
The first GIN layer generated an updated feature representation, which was then passed to the second GIN layer. Here, we introduced residual connections, which added the output of the first GIN to the output of the second GIN. These residual connections helped address the vanishing gradient problem and enabled the network to learn more complex features. The output of the second GIN was then passed to the final GIN layer through a second residual connection, which added the output of the initial input
The R2eGIN was constructed with 3 message-passing layers to obtain graph-level embeddings

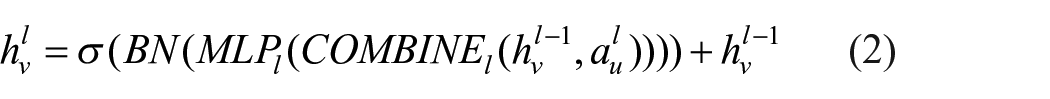
Here,

The node embeddings

where
Figure 2 illustrates how information was propagated and aggregated within the R2eGIN layers.
22
Information from the nodes was gathered through a message-passing process, where this neighbor information, represented as

Node-edge neighbor aggregation and node-neighborhood combination within R2eGIN.
Residual learning
In a GNN architecture designed for molecular tasks, shallow models typically failed to effectively capture key information and interactions within molecular data graphs. Meanwhile, as depth increased, models encountered the vanishing gradient problem, leading to performance degradation and suboptimal results.27,28 To mitigate these issues, a residual approach was used. R2eGIN extended the GIN layer with 2 stacked layers by applying residual connections.
Figure 3 shows the block structure of R2eGIN, where


Structure of the R2eGIN.
Reconstruction blocks
The implementation of the reconstruction block aimed to compel the model to learn rich and informative representations that could be used to reconstruct the original node features, thereby enhancing the model’s understanding of molecular structures. We incorporated a reconstruction block, consisting of 2 fully connected layers, to reconstruct the original node features from the graph representations. In addition, we included a prediction block, comprising a single fully connected layer followed by a sigmoid activation function, to predict the target. Thus, enabling the model to learn improved node feature representations and enhance prediction performance. The total loss was calculated as follows

Where

With
To predict PARPi, R2eGIN used a prediction block consisting of a single fully connected layer followed by a sigmoid activation function. Similar to the reconstruction layer, the graph-level molecular representation
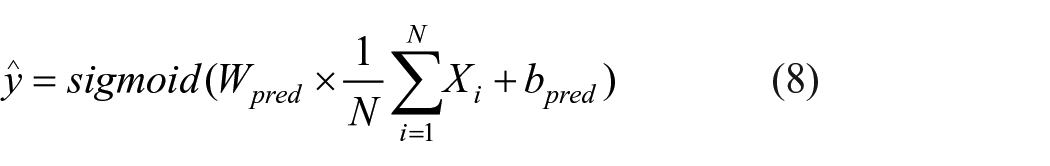
where
Molecular graph embedding
The transformation of molecular representations began by converting Simplified Molecular Input Line Entry Specification (SMILES) strings into molecular graph structures, where nodes represent atoms and edges represent chemical bonds. To obtain rich and informative molecular representations, we used crucial features, as employed by literatures29-32 to construct an accurate model. As presented in Table 1, we incorporated 7 atomic features and 2 edge features, including Bond type (BT), Bond direction (BD), Atom type (AT), Atom Formal charge (AF), Atom Chirality (AC), Atom Hybridization (AH), Number of hydrogen atoms bonded (HA), Implicit valence (AV), and Atom Degree (AD). Molecular structures inherently involve complex atomic interactions and electronic structures, and bond features encapsulate rich information regarding molecular frameworks and conformational isomers. Embedded molecular graphs can implicitly capture essential molecular information and inter-atomic interactions, while providing insights into the side characteristics of molecular bonds. The input edges and nodes for aggregation can be formalized by

Details of atom and edge features.

Experimental
Data sets
The data set employed for the PARPi study was initially procured from various publicly accessible sources, BindingDB, 33 PubChem,34,35 and ChEMBL. 36 In this study, we accessed the experimental data set published by Ai et al. 37 The data set comprises 4 PARP isoform data sets: PARP-1 (UniProt ID: P09874), PARP-2 (Q9UGN5), PARP-5A (O95271), and PARP-5B (Q9H2 K2). The detailed distribution of activity labels within each data set is as follows: PARP-1 (3119 active, 658 inactive), PARP-2 (271 active, 141 inactive), PARP-5A (702 active, 147 inactive), and PARP-5B (628 active, 104 inactive), as depicted in Figure 4A. Overall, the distribution of compound activity labels exhibited an imbalance, with a total of 4720 active and 1050 inactive compounds across all data sets. The average percentage of active and inactive labels for the entire data set is 20.8% and 79.2%, respectively (Figure 4B). In addition, chemical space analysis was conducted on the compounds within the PARP-1 (Figure 4C), PARP-2 (Figure 4D), PARP-5A (Figure 4E), and PARP-5B (Figure 4F) data sets, using molecular weight (MW) and AlogP as defining parameters. The analysis revealed a broad molecular weight range, spanning from 121.139 to 725.683, and the AlogP range of −1.946 to 8.700. These findings indicated that the compounds present in the modeling data sets encompassed a diverse chemical space.

Compound distribution and chemical space analysis of PARP datasets.
Evaluation metrics
We evaluated the performance of R2eGIN on PARPi prediction using accuracy (ACC.), balanced accuracy (BA) Matthews correlation coefficient (MCC), F1-score (F1), precision (Prec.), and area under the ROC curve (AUC). Given false positive (FP), true positive (TP), false negative (FN), and true negative (TN), their formulas are as follows:





Experimental settings
Experiments were conducted using PyTorch on Ubuntu 18.0.4 64bit with NVIDIA GTX 1080Ti × 4 and Intel Core i9-9900K (3.60 GHz). We trained R2eGIN in 200 epochs using the Adam optimizer with a learning rate of 0.001. A specific model with a 3-layer GIN acted as the R2eGIN backbone. The dropout rate range was (0.2, 0.5), and batch sizes were (64, 128, 512) as detailed in Table 2.
Hyperparameter settings.

Results
R2eGIN for PARPi prediction
R2eGIN achieved the highest accuracy (0.926), MCC (0.728), F1 score (0.956), and BA (0.836) on the PARP-1 data set as presented in Table 3. These results highlighted the model’s ability to effectively capture both local and global structural information within molecular graphs. In addition, the use of residual connections and reconstruction block integrated background information from the initial embeddings into the propagation layers, enabling the model to learn effectively without losing critical information necessary for PARPi classification.
Performance of R2eGIN.

Comparison with existing models
To evaluate the effectiveness of R2eGIN, we compared diverse graph learning methods, including Chemprop, 38 MPNN, 26 FPGNN, 37 AttentiveFP, 39 GCN, 40 GAT, 25 and GIN. 24 Overall, the proposed model demonstrated superior performance, exhibiting average scores of 0.873 for ACC, 0.576 for MCC, 0.919 for F1, and 0.748 for BA, as shown in Figure 5A. Concurrently, Chemprop secured the second-highest position, achieving an average ACC of 0.848 with a standard deviation (STD) of 0.041 and an average F1-score of 0.904 with a STD of 0.039. FPGNN registered the second-best average BA of 0.710, while GIN ranked as the second highest in average MCC, with a score of 0.466 (Table 4).

Comparative analysis of performance between the R2eGIN and the other methods.
Comparison performance on average scores between R2eGIN and the other models on 4 data sets PARPi.

The best results are highlighted in
The comparative evaluation of R2eGIN and eight baseline models, performed on the test sets of PARP-1, PARP-2, PARP-5A, and PARP-5B, is visualized in Figure 5B. The FPGNN model combined the strengths of traditional molecular fingerprint representations with GNN. While this model performed well on specific tasks, 41 the use of 2 distinct representations could lead to feature redundancy. Certain atomic properties already represented by molecular descriptors might be redundantly incorporated into the graph’s feature representation, potentially diminishing the overall efficiency and clarity of the representation. GAT and AttentiveFP implemented attention mechanisms, enabling the models to dynamically assign importance weights to neighboring nodes within the graph representation. Although this approach effectively enhanced the model’s ability to focus on relevant graph components, it is crucial to acknowledge that no single model is universally optimal across all tasks. Meanwhile, Chemprop model’s superior performance on the PARP-2 data set could be attributed to the data set’s unique characteristics, which might exhibit a molecular structure distribution and activity patterns particularly well-suited to Chemprop capabilities, as detailed in Table 5. Conversely, for the PARP-5A data set, FPGNN (BA 0.681) and GIN (ACC 0.871; MCC 0.595; F1 0.927) outperformed Chemprop. Similarly, on the PARP-5B data set, AttentiveFP (MCC 0.420) and GCN (ACC 0.879; F1 0.933; BA 0.644) ranked second only to the proposed R2eGIN model. Overall, a comparative analysis of the eight models for predicting PARP inhibitor activity revealed intriguing patterns across different PARP isoform data sets. The findings demonstrated that the R2eGIN model consistently surpassed other models in terms of ACC, MCC, F1, and BA on the PARP-1, PARP-5A, and PARP-5B data sets, highlighting the robust performance of the proposed model in PARPi prediction tasks.
Overall performance comparisons of different PARPi prediction models.

The best results are highlighted in
Ablation study
We performed an ablation study to assess the performance improvement gained by incorporating 2 components, residual connection and the reconstruction block, in the proposed method. Four experimental scenarios were designed: M1, M2, M3, and M4, as presented in Table 6. In scenario M1, the GIN model was used without residual learning or the reconstruction block. Scenario M2 features the GIN backbone with residual connections but without the reconstruction block. In scenario M3, the GIN backbone is enhanced with the reconstruction block but without residual connections. Finally, scenario M4 corresponded to the proposed R2eGIN model. The ablation experiment demonstrated that R2eGIN outperformed the other scenarios across evaluation metrics as shown in Figure 6.
Scenario and abbreviations used in ablation study.


Performances of different model variants in ablation experiment.
Conclusion
In this study, we present the R2eGIN model, which is GIN by incorporating residual connection and reconstruction network. The experimental results on 4 PARPi data sets (PARP-1, PARP-2, PARP-5A, and PARP-5B) demonstrate that the R2eGIN model derives superior performance over state-of-the-art models on the task of PARPi prediction. This study is the first to demonstrate that a GIN-based model enhanced with residual and reconstruction mechanisms is well-suited for predicting PARP inhibitors. In addition, it reveals key strategies for screening potential molecules and accelerating virtual screening methods in drug discovery, particularly for cancer therapy development. Although R2eGIN demonstrates good performance and potential in predicting PARP inhibition activity, niche for improvement for future research is available. The performance of R2eGIN is constrained by the quality and quantity of available data, particularly in the PARP-2 data set, consisting of only 412 samples, with 271 actives and 141 inactives. Overall, across the 4 data sets, inactive molecular data accounts for an average of only 20.8%, while active molecular data constitutes 79.2%. Therefore, to enhance the reliability and robustness of PARPi predictions, we will continue refining our methodology by advancing molecular representation through next-generation deep learning models, integrating GNN with complementary computational approaches to optimize performance, 42 and meticulous collection and curation of high-quality data. In addition, we prioritize improving model interpretability, as it is essential for gaining deeper insights into model behavior.
Footnotes
Author Contributions
Conceptualization: Candra Zonyfar, and Jeong-Dong Kim; Methodology: Candra Zonyfar, and Jeong-Dong Kim; Software: Candra Zonyfar, and Soualihou Ngnamsie Njimbouom; Validation: Soualihou Ngnamsie Njimbouom, Sophia Mosalla, and Jeong-Dong Kim; Formal analysis: Candra Zonyfar, Soualihou Ngnamsie Njimbouom, and Jeong-Dong Kim; Investigation: Candra Zonyfar, Sophia Mosalla, and Jeong-Dong Kim; Resources: Candra Zonyfar, Soualihou Ngnamsie Njimbouom, Sophia Mosalla, and Jeong-Dong Kim; Data curation: Candra Zonyfar, Soualihou Ngnamsie Njimbouom, and Sophia Mosalla; Writing – original draft: Candra Zonyfar; Writing – review & editing: Candra Zonyfar, Soualihou Ngnamsie Njimbouom, and Jeong-Dong Kim; Visualization: Candra Zonyfar, Soualihou Ngnamsie Njimbouom, and Jeong-Dong Kim; Supervision: Jeong-Dong Kim; Project administration: Jeong-Dong Kim; Funding acquisition: Jeong-Dong Kim. All authors have read and agreed to the published version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the MSIT (Ministry of Science ICT), Korea, under the National Program for Excellence in SW, supervised by the IITP (Institute of Information & Communications Technology Planning & Evaluation) in 2025 (No. 2024-0-00023).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.