
Abstract
Transposable elements (TEs) or transposons are thought to play roles in animal physiological processes, such as germline, early embryonic, and brain development, as well as aging. However, their roles have not been systematically investigated through experimental studies. In this study, we created a catalog of genes directly involved in replication, excision, or integration of transposon-coding DNA, which we refer to as transposon DNA processing genes (TDPGs). Specifically, to bridge the gap to experimental studies, we sought potentially functional TDPGs which maintain intact open reading frames and the amino acids at their catalytic cores on the latest long-read genome assembly of Caenorhabditis elegans, VC2010. Among 52 519 TE loci, we identified 145 potentially functional TDPGs encoded in long terminal repeat elements, long interspersed nuclear elements, terminal inverted repeat elements, Helitrons, and Mavericks/Polintons. Our TDPG catalog, which contains a feasible number of genes, allows for the experimental manipulation of TE mobility in vivo, regardless of whether the TEs are autonomous or non-autonomous, thereby potentially promoting the study of the physiological functions of TE mobility.
Introduction
Transposable element (TE)-related repetitive DNA elements occupy significant proportions of eukaryotic genomes: 12% in Caenorhabditis elegans, 4% to 17% in Drosophila melanogaster, 37% in mouse, 46% in human, and ~85% in corn.1,2 Transposable elements are thought to play roles in animal physiological processes, such as germline and early embryonic and brain development, 3 as well as aging. 4 However, experimental investigations through the systematic manipulation of TE activity have rarely been performed. Due to the large number of TEs present in any given genome, it is necessary to devise new approaches for their systematic manipulation.
TEs transpose through enzymes encoded within their own regions. Depending on the medium required for transposition, TEs are primarily classified into retrotransposons (class I) and DNA transposons (class II), and these include both autonomously mobile and non-autonomously mobile TEs.5-7 Autonomously mobile class I and II TEs are further subdivided by the transposition mechanism 6 into the 4 retrotransposon orders: the long terminal repeat (LTR) elements, long interspersed nuclear elements (LINEs), Dictyostelium intermediate repeat sequences (DIRS), and Penelope-like elements (PLEs) as well as the 4 DNA transposon orders: the terminal inverted repeat (TIR) elements, Helitrons, Maverick/Polintons (MP), and Cryptons. Regarding retrotransposons, LTR elements transpose by generating DNA copies from a RNA transcript via reverse transcriptase (RT). Integrase (IN) is required to efficiently insert LTR element DNA fragments into new genomic loci. Similarly, LINEs transpose by replicating DNA copies from a transcript via LINE-specific RT. In this case, reverse transcription starts at the 3′-end of the nick site on the genomic DNA generated via the Endonuclease (EN) domain at the N-terminus of the RT. Dictyostelium intermediate repeat sequence transposition occurs when free circular double-stranded DNAs are generated via reverse transcription. The circular double-stranded DNAs are then integrated into other loci by Tyrosine-Recombinase (Tyr-REC).8-11 In PLE transposition, the DNA fragment of PLE is reverse-transcribed from the 3′ end of the nick site on the genome generated by the GIY-YIG EN activity in the PLE-specific RT. 12 As for DNA transposons, the TIR elements transpose via the EN activity of Transposase (TP). The TP recognizes TIRs at both ends of TIR element and cleaves double-stranded DNA (dsDNA) to generate a free DNA fragment. This fragment is integrated into another genomic locus by the EN activity of TP.13-15 Helitron transposition occurs when the Helitron DNA element is nicked via the EN activity of the REP domain in REP-Helicase (REP-HEL) and is then unwound into single-stranded DNA (ssDNA) through HEL activity. Although it remains controversial whether the Helitron ssDNA is replicated before integration into another genomic locus, a free Helitron fragment is integrated into another locus by EN activity of the REP domain.16-20 In MPs, free DNA copies are replicated by DNA Polymerase B encoded within MP. These MP DNA copies are inserted into other genomic loci by MP-encoded INs.21-24 Cryptons transpose after being excised as free circular DNAs (circDNAs) by Tyr-REC. The free circDNAs are integrated into another genome locus by Tyr-REC.11,25
As described above, the transposition of TEs can be conceptualized as occurring through 2 DNA processing reactions: (1) the production of a free TE DNA and (2) its integration into another genomic locus. In retrotransposons LTR element and DIRS, and DNA transposon MP, the free TE DNA is generated and integrated by different enzymes. In retrotransposons LINE and PLE, and DNA transposon Helitron, these processes are mediated by different enzymatic activities associated with each functional domain in the same enzyme. In DNA transposons TIR element and Crypton, these processes are mediated by the same functional domains in the same enzyme. Hereafter, we refer to the genes involved in the production and/or integration of free TE DNAs as transposon DNA processing genes (TDPGs).
Detailed analyses of the open reading frame (ORF) structure and amino acid sequence of TDPGs have been primarily focused on individual TE orders.26-33 These sequence analyses alone offer limited insights into their roles in TE mobile activity. For instance, a TDPG containing numerous mutations or deletions may still retain enough catalytic activity to facilitate transposition in vivo. In this study, we have cataloged potentially functional TDPGs, which maintain the ORF structure and the conserved amino acids at the catalytic core across all representative TE orders in a single species, C. elegans. This work provides a foundation for systematically investigating the physiological roles of TE mobility in C. elegans, paving the way to seek conserved mechanisms across different organisms.
Methods
Bioinformatic analysis
The VC2010 genome assembly was downloaded from https://www.ebi.ac.uk/ena/browser/view/UNSB01. To identify TEs, RepeatMasker version 4.1.0 (http://www.repeatmasker.org/) was used with options -no_is for skipping bacterial insertion element check, -s: slow search for more sensitivity, and -pa 8 for sequencing batch jobs to run in parallel. For RepeatMasker analysis, we used Dfam_3.1 library. 34 To identify ORFs, SNAP (Semi-HMM-based Nucleic Acid Parser), version 2006-07-28, 35 was used to analyze a FASTA file containing all the repeat sequences identified by RepeatMasker. The command used was snap HMM/C.elegans.hmm TEgenomeseq.fasta. Here, C.elegans.hmm contained parameters optimized for the C. elegans genome, and TEgenomeseq.fasta contained all the repeat sequences identified by RepeatMasker. To infer the function of proteins encoded in ORFs, DIAMOND (Double Index AlignMent Of Next-generation sequencing Data) (v2.0.2.140) 36 blastx tool was used to compare our ORF sequences identified by SNAP against the UniRef50 protein database. Default parameters were employed, with no additional sequence masking or complexity adjustments. The search sensitivity was set to the default “fast” mode. The following command line was used for the search: diamond blastx –db uniref50.fasta –query TDPGsORF.fasta –out blastx.txt –outfmt 6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen. Here, TDPGsORF.fasta contained ORF sequences identified by SNAP. No specific e-value threshold was set; hence, the default cutoff of 0.001 was applied. UniRef50 protein ID produced by DIAMOND was translated into functional protein IDs using the website Uniprot (https://www.uniprot.org/uploadlists/). To align amino acid sequences, MAFFT v7.45337,38 was used. A sequence alignment viewer was downloaded from https://github.com/dmnfarrell/teaching/blob/master/pyviz/bokeh_sequence_align.ipynb.
Results
Bioinformatic identification of TDPGs in the C. elegans genome
We first applied RepeatMasker, an algorithm that identifies TEs (https://www.repeatmasker.org/), to the latest C. elegans VC2010 genome assembly (for the detail, see Materials and Methods). The VC2010 genome assembly was constructed using long-read sequencing technology. 39 Long-read sequencing helps to accurately determine the genomic locations of repeat elements, such as TEs, and thereby results in more accurate estimates of the copy numbers of TEs in the genome, which is challenging for short-read sequencing. Our RepeatMasker analysis showed that repeat elements occupy 13.86% of the C. elegans genome (Supplementary Table 1). This result is consistent with previous reports that repetitive elements comprise 12% to 17% of the C. elegans genome.28,40,41 After excluding simple, satellite, and low-complexity repeat elements, we identified 52 519 TE loci (Supplementary Table 1). To identify protein-coding elements among these 52 519 TE copies, we used an ab initio gene finder, SNAP. 35 The SNAP analysis identified 808 potential gene-coding regions. Among them, 428 genes conserved the complete ORF structure. To infer the function of genes encoded in these complete ORFs, we used DIAMOND, an algorithm for fast and sensitive protein alignment. 36 DIAMOND identified 80 RT, 11 IN, 189 TP, and 5 REP-HEL genes, for a total of 285 TDPGs (for the detail, see Materials and Methods). In following sections, we systematically dissected these genes to create a curated list of potentially functional genes.
TDPGs in LTR elements in C. elegans
Among the 80 RT genes, 19 genes were encoded in LTR elements (rtz_LTRs) and 61 genes were encoded in LINEs (rtz_LINEs) (Supplementary Tables 2 and 4). Reverse transcriptases, ie, RNA-dependent DNA polymerases, have 5 evolutionarily conserved motifs (A, B′, C, D, and E).42,43 Asp residues in Motifs A and C are widely conserved in all RNA/DNA-dependent DNA polymerases and DNA/RNA-dependent RNA polymerases. 44 Previous crystal structure analyses showed that 1 Asp in Motif A and 2 Asp residues in Motif C form a catalytic triad for holding 2 bivalent metal ions for conjugating the alpha phosphate of a new dNTP to the OH group to the 3′ end of the DNA strand.45-47 Mutations in Asp residues of Motifs A or C reportedly abolish RT activity.48-51 By aligning RTZ_LTRs with the reference RTs, we identified 8 RTZ_LTRs with conserved Asp residues in Motifs A and C (red asterisks in Figure 1A and Table 1). In addition, these 8 RTZ_LTRs preserved (1) the conserved Gly residue in Motif B′, 52 which interacts with the incoming nucleotide and template strand, 53 and (2) the Leu and Gly residues in Motif E, which fixes the primer strand and positions it toward the active site 53 (Figure 1C and Table 1).

Potentially functional rtz_LTRs and inz_LTRs encoded in long terminal repeat (LTR) elements. (A) Alignment of catalytic core domains of RTZ_LTRs with reference Reverse transcriptases (RTs). Abbreviations: HIV-1: Chain C, HIV-1 RT P66 subunit of human immunodeficiency virus type 1 [5TXO_C], Hep-B: Hepatitis B virus RT_like family [QFR04538], ERV-K: Pol protein of human endogenous retrovirus K [CAA76882], RSV: Pol of Rous sarcoma virus [CAA48535]. (B) Alignment of catalytic core domains of INZ_LTRs with reference Integrases (INs). Abbreviations: ASV: IN in avian sarcoma virus [1ASU_A], HERVK: Pol protein in human endogenous retrovirus K [CAA76885], MMLV: p46 IN in Moloney murine leukemia virus (MoMLV) [NP_955592.1], HIV: IN in human immunodeficiency virus type 1 [1BIZ_A], Gypsy_Dm: IN, Gypsy endogenous retrovirus in Drosophila melanogaster [CAB69645], TEDV_Tm: ORFB in TED virus in Trichoplusia ni [YP_009507248], TP1731: Pol polyprotein in transposon_1731 in D. melanogaster [S00954], RVRP_At: retrovirus-related like polyprotein in Arabidopsis thaliana [CAB78488_1], Copia_Dm: Gag-Int-Pol protein in COPIA in D. melanogaster [P04146], Ty-3_Sc: Gag-Pol polyprotein in Ty3-G in Saccharomyces cerevisiae [GFP69998.1]. Asterisks indicate conserved residues. Red asterisks indicate residues for identifying potentially functional TDPGs. (C) Genomic positions of 8 potentially functional rtz_LTRs and 7 potentially functional inz_LTRs in the Caenorhabditis elegans genome. Gray lines represent chromosomes. Red circles over and under chromosome indicate positions of potentially functional genes of rtz_LTR-n and inz_LTR-n, respectively. Numbers over and under chromosome indicate numbers of rtz_LTR and inz_LTR genes, respectively. Vertical ticks and × marks with vertical ticks over and under chromosome indicate incomplete and complete ORFs, respectively, of rtz_LTRs and inz_LTRs, respectively.
List of potentially functional rtz_LTRs and inz_LTRs.

Evolutionarily conserved amino acids within the Reverse transcriptase (“RT”) and Integrase (“IN”) within potentially functional RTZ_LTRs and INZ_LTRs. The consecutive amino acid single letters without spaces indicate conserved amino acids within the same domain. See amino acids indicated by asterisks in Figure 1A and B. A dash (-) indicates the absence of an ID or conserved amino acid. The “Class” denotes TE identity as determined by RepeatMasker analysis. Chromosome ID (“Chr”) and genomic positions (“Position”) are provided for each gene.
In RTZ_LTR-11, RTZ_LTR-17, and RTZ_LTR-19, the second Asp residue in Motif C was substituted with Asn (black asterisk in Figure 1A and Table 1). As described earlier, the second Asp residue is involved in a catalytic triad. Site-directed mutagenesis at the second Asp residue, including mutagenesis to Asn as found in RTZ_LTR-11, RTZ_LTR-17, and RTZ_LTR-19, significantly reduces RT activity in human immunodeficiency virus (HIV).48-51 However, amino acid substitution at the second Asp to Asn has been found in functional RNA-dependent RNA polymerases in negative-strand RNA viruses.42,44,54 Therefore, we considered the possibility that RTZ_LTR-11, RTZ_LTR-17, and RTZ_LTR-19 might still be functional.
In RTZ_LTR-11 and RTZ_LTR-19, the conserved Gly and Lys in Motif D were substituted. In RTZ_LTR-3, RTZ_LTR-8, and RTZ_LTR-17, the conserved Gly in Motif D was substituted. Amino acid substitutions at Gly in Motif D are often observed in RNA-dependent RNA polymerases in negative-strand RNA viruses. 42 Therefore, we considered the possibility that RTZ_LTR-3, RTZ_LTR-8, and RTZ_LTR-17 might still be functional. On the other hand, Lys in Motif D is highly conserved throughout polymerases.42,43 Motif D functions for forming a phosphodiester bond with dNTPs with the 3′-OH (hydroxyl) of the primer.55,56 Motif D in RTZ_LTR-11 and RTZ_LTR-19 may be nonfunctional. Nevertheless, given the conservation of the 2 critical amino acids holding 2 bivalent metal ions, and the fact that we did not perform functional testing, we include RTZ_LTR-11 and RTZ_LTR-19 as potentially functional TDPGs. Taken together, these results led us to classify all 8 rtz_LTRs as potentially functional genes (Table 1 and Supplementary Table 2).
Next, we identified 11 IN genes encoded in LTR elements (inz_LTRs) (Supplementary Table 3). The catalytic core domain of IN has an evolutionarily conserved DD35E motif that is required for EN activity.57-59 DD35E holds 2 metal ions required for the catalysis involved in integrating a free DNA fragment into the genome.60,61 Amino acid substitution at the 3 critical amino acid residues in the DD35E motif abolishes EN activity of INs in Rous sarcoma virus (RSV) and HIV.57-59,62 By aligning the 11 INZ_LTRs with the reference INs, we identified 7 INZ_LTRs with conserved DD35E triads (Figure 1B, Table 1 and Supplementary Table 3). Therefore, we considered these 7 inz_LTRs as potentially functional genes. Potentially functional RT and IN genes generally co-occurred in LTR element loci, except for the loci encoding rtz_LTR-11 and rtz_LTR-17, which lacked the corresponding inz_LTR, and the locus encoding inz_LTR-10, which lacked the corresponding rtz_LTR (Figure 1C and Table 1).
TDPGs in LINEs in C. elegans
By aligning the 61 remaining RTs (from the initial 80 RTs, excluding the 19 RTs analyzed as RTZ_LTRs in the previous section) with reference RTs, we identified 28 RTZ_LINEs that had Asp residues conserved in Motifs A and C (red asterisks in Figure 2A and Table 2). In addition, these 28 RTZ_LINEs had residues conserved in Motifs B′ and C (black asterisks in Figure 2A and Table 2). These 28 RTZ_LINEs had Lys but not Gly residues conserved in Motif D. Amino acid substitution of Gly in Motif D is often observed in RNA-dependent RNA polymerases in negative-strand RNA viruses. 42 Therefore, we held out the possibility that the RTZ_LTRs with amino acid substitution of Gly in Motif D are functional. In addition, in RTZ_LINE-61, Gly residues in Motif E were substituted. The substitution in this residue was found in RT in HIV-1 and Hepatitis B virus (HepB), 42 leading us to consider the possibility that RTZ_LINE-61 is potentially functional (black asterisks in Figure 2A and Table 2). In summary, analysis of RT domains suggests that these 28 rtz_LINEs are potentially functional.
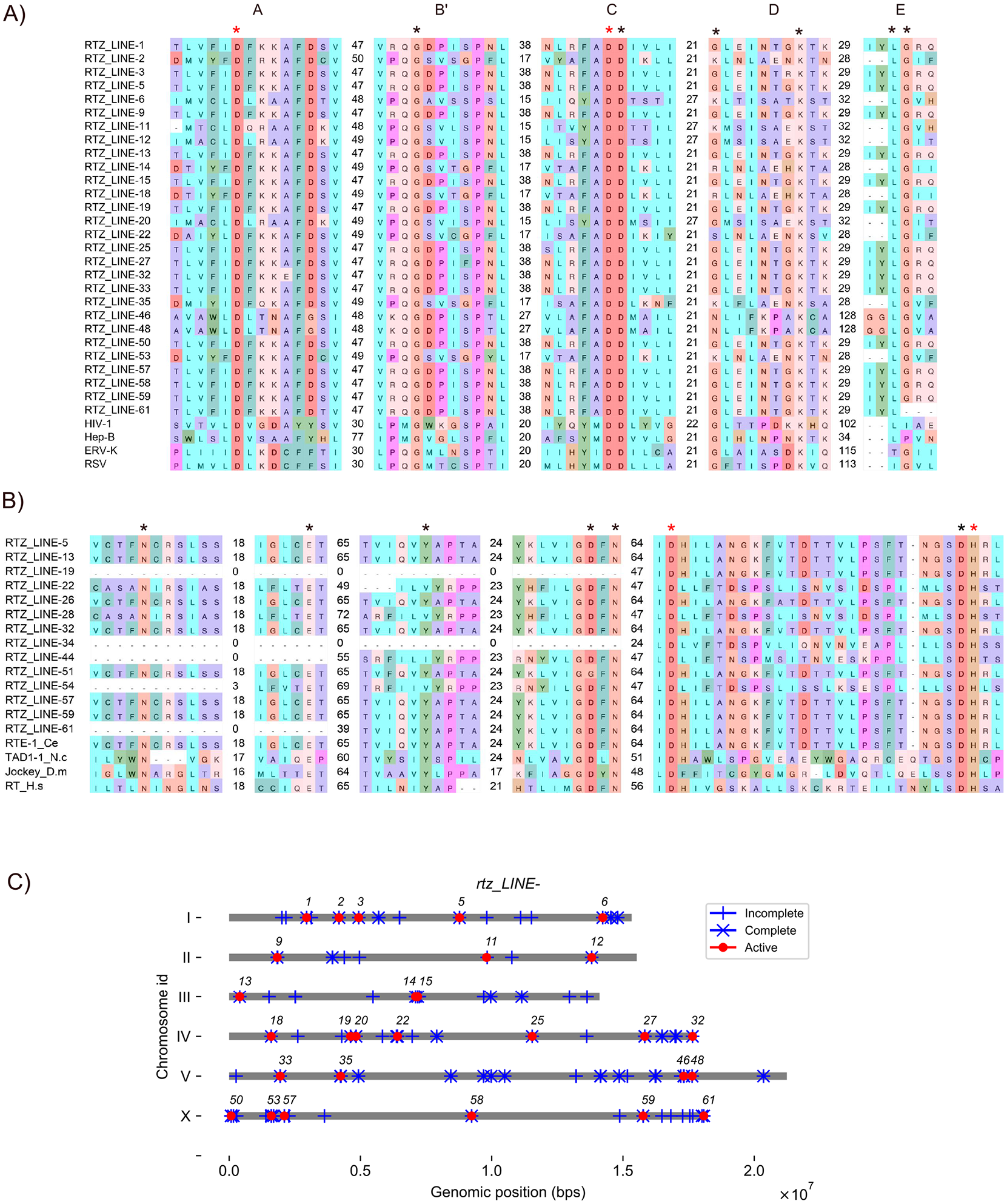
Potentially functional rtz_LINEs encoded in long interspersed nuclear elements (LINEs). (A) Alignment of reverse transcriptase (RT) domains of RTZ_LINEs with reference RTs. Abbreviations: HIV-1: Chain C, HIV-1 RT P66 subunit of human immunodeficiency virus type 1 [5TXO_C], Hep-B: hepatitis B virus RT_like family [QFR04538], ERV-K: Pol protein of human endogenous retrovirus K [CAA76882], RSV: Pol of Rous sarcoma virus [CAA48535]. (B) Alignment of the Endonuclease (EN) domains of RTZ_LINEs with reference ENs. Abbreviations: RTE-1_Ce: apurinic-apyrimidic EN domain containing RT of non-LTR retrotransposon in Caenorhabditis elegans [AAC72298.1], AP_End_Hs: DNA-(apurinic or apyrimidinic site) EN in Homo sapiens [NP_542379.1], TAD1-1_N.c: Exonuclease-Endonuclease-Phosphatase (EEP) domain containing Pol protein Neurospora crassa [AAA21781.1], Jockey_D.m: EEP domain containing RT in Drosophila melanogaster [AAA28675.1], RT_H.s: EN domain containing RT of L1, H. sapiens [AAB59368.1]. Asterisks indicate conserved residues. Red asterisks indicate residues for identifying potentially functional TDPGs. (C) Genomic positions of 28 potentially functional rtz_LINEs in the C. elegans genome. Gray lines represent chromosomes. Red circles indicate positions of potentially functional genes of rtz_LINE-n. Numbers indicate numbers of rtz_LINE-n genes. Vertical ticks and × marks indicate incomplete and complete ORFs, respectively, of rtz_LINEs.
List of potentially functional rtz_LINEs.

Evolutionarily conserved amino acids within the Reverse transcriptase (“RT”) and Endonuclease (“EN”) domains within potentially functional RTZ_LINEs. The consecutive amino acid single letters without spaces indicate conserved amino acids within the same domain. See amino acids indicated by asterisks in Figure 2A and B. A dash (-) indicates the absence of a conserved amino acid. The “Class” denotes TE identity as determined by RepeatMasker analysis. Chromosome ID (“Chr”) and genomic positions (“Position”) are provided for each gene.
Next to study the EN domain, we aligned the 61 RTZ_LINEs with reference ENs. A previous experimental test of transposition activity in 138 human L1 copies revealed that the Asp and His residues are essential, but conserved amino acid residues in other motifs tolerate multiple mutations. 63 We identified 14 RTZ_LINEs in which the critical Asp and His residues were conserved in their EN domains (red asterisks in Figure 2B and Table 2). Among the 14 RTZ_LINEs, RTZ_LINE-19 and RTZ_LINE-34 lacked a large N-terminal portion of the EN domain, whereas RTZ_LINE-44 and RTZ_LINE-61 lacked about half of this portion (Figure 2B). Nevertheless, in the absence of experimental tests of the function of N-terminal deletion EN mutants, we held out the possibility that all the 14 RTZ_LINEs have potentially functional EN domains. Taken together, we concluded that 6 rtz_LINEs encoded both potentially functional RT and EN domains (rtz_LINE-5, rtz_LINE-13, rtz_LINE-22, rtz_LINE-57, rtz_LINE-59, and rtz_LINE-61; Figure 2A and B, Table 2 and Supplementary Table 4). However, it has been noted that human L1 can transpose via an EN-independent, RT-dependent mechanism. 64 Therefore, we concluded that the 28 rtz_LINEs with conserved RT domains (Figure 2A and Table 2) are potentially functional genes for LINE transposition. The 28 rtz_LINEs distributed across each chromosome (Figure 2C) and amino acid sequences of these genes could be grouped into 4 homologous clusters (Supplementary Figure 1 and Supplementary Table 4).
TDPGs in TIR elements in C. elegans
Terminal inverted repeat element is composed of 19 super families: hAT, Tc1/mariner, CACTA (En/Spm), Mutator (MuDR), P, PiggyBac, PIF/Harbinger, Mirage, Merlin, Transib, Novosib, Rehavkus, ISL2EU, Kolobok, Chapaev, Sola, Zator, Ginger, and Academ. 65 The transposition of TIR elements is mediated by Transposase (TP), which has a conserved DDD/E motif at the catalytic core. 65 Structural analysis suggests that the DDD/E motif holds 2 metal ions to cleave and integrate dsDNA of the TIR element.66-68 Mutation of the conserved DDD/E motif abolishes TP activity.69,70 By aligning the 189 TPZs with reference TPs, we identified 94 TPZs that had DDD/E motifs conserved that were homologous to those of Tc1/mariner family TPs (Figure 3A and Supplementary Table 5). No TP aligned to DDD/E motifs of other TP super families. Thus, we considered these 94 tpzs to be potentially functional genes (Supplementary Table 5). tpzs were distributed across each chromosome (Figure 3B) and amino acid sequences of these genes predominantly formed 3 main homologous clusters (Supplementary Figure 2 and Supplementary Table 5).

Potentially functional tpzs encoded in terminal inverted repeat (TIR) elements. (A) Alignment of catalytic core domains of TPZs with reference Transposases (TPs). Amino acid sequences of Pogo_SM, CIRT2_CA, and Mar1_TV as examples of Tc1/mariner class TPs were arbitrarily obtained from Yuan and Wessler. 65 Red asterisks indicate conserved residues used to identify potentially functional TDPGs. (B) Genomic positions of 94 potentially functional tpzs in the Caenorhabditis elegans genome. Gray lines represent chromosomes. Red circles indicate positions of potentially functional genes of tpz-n. Numbers indicate numbers of tpz genes. Vertical ticks and × marks indicate incomplete and complete ORFs, respectively, of tpzs.
TDPGs in Helitrons in C. elegans
Helitron transposition is mediated by a SF1 family Helicase (HEL) with REP domain (REP-HEL). The 4 SF family HELs (SF1, SF2, SF3, and SF4) have conserved Motif I and Motif II domains. Motifs I and II correspond to the Walker A and Walker B domains, respectively, which are widely conserved among NTP-binding proteins.71,72 The Walker A/Motif I and Walker B/Motif II domains exhibit conservation of the Lys and Asp-Glu residues, respectively.72-75 Crystallographic analysis showed that the Lys in Motif I contacts a magnesium ion and β phosphate of ATP and functions to stabilize the transition state during ATP hydrolysis. Asp-Glu residues in Motif II are also involved in ATP hydrolysis.76,77 Mutations at the Lys in Motif I or Asp-Glu in Motif II abolish HEL activity.78-81
By aligning the 5 REP-HELs (RHZs) with reference HEL domains, we identified 5 RHZs that had Lys residues conserved in Motif I and had Asp-Glu residues conserved in Motif II (red asterisks in Figure 4A and Table 3). In addition, these 5 RHZs had (1) a conserved residue in Motif Ia (black asterisks in Figure 4A and Table 3), which functions for ssDNA binding and energy transfer from the ATP-binding site to the DNA-binding site 76 ; (2) conserved Gly-Asp and other resides in Motif III (black asterisk in Figure 4A and Table 3), which is involved in contacting nucleotide γ-phosphates 76 ; (3) a conserved Arg residue in Motif IV, which may be involved in NTP hydrolysis 76 ; (4) a conserved residue in Motif IV/V, the function of which is not well understood 18 ; (5) conserved residues in Motif V, which interacts with the sugar-phosphate backbone of DNA 76 ; and (6) conserved residues in Motif VI, which may form part of the ATP-binding cleft to couple ATPase activity to HEL activity 76 (Figure 4A and Table 3). Therefore, we considered these 5 rhzs to encode potentially functional HEL domains.

Potentially functional rhzs encoded in Helitrons. (A) Alignment of amino acid sequences of helicase (HEL) domains with reference HELs. PIF1 in Saccharomyces cerevisiae [P07271], HEL_T4 in Enterobacteria phage T4 [P32270], TraA Sinorhizobium fredii [P55418], TraI_EC Escherichia coli [P14565], TRWC E. coli [Q47673]. (B) Alignment of amino acid sequences of REP domains with reference REPs. Rep_Bb in Brevibacillus borstelensis [BAA07788.1], Rep plasmid pVT736-1 [AAC37125.1], Rep_Pf3P. phage Pf3 [AAA88392.1], Rep_EC IS91 [BCN22733.1], TnpA_EC IS91 TnpA E. coli [QIC00531.1]. Asterisks indicate conserved residues. Red asterisks indicate residues used for identifying potentially functional TDPGs. (C) Genomic positions of 5 potentially functional rhzs in the Caenorhabditis elegans genome. Gray lines represent chromosomes. Red circles indicate positions of potentially functional genes of rhz-n. Numbers indicate numbers of rhz genes. Vertical ticks and × marks indicate incomplete and complete ORFs, respectively, of rhzs.
List of potentially functional rhzs.

Evolutionarily conserved amino acids within the Helicase (“Hel”) and Endonuclease (“REP”) domains within potentially functional RHZs. The consecutive amino acid single letters without spaces indicate conserved amino acids within the same domain. See amino acids indicated by asterisks in Figure 4A and B. A dash (-) indicates the absence of a conserved amino acid. The “Class” denotes the TE identity as determined by RepeatMasker analysis. Chromosome ID (“Chr”) and genomic positions (“Position”) are provided for each gene.
In the REP domain, the HUH Y2 motif (in which U is a hydrophobic residue) is evolutionarily conserved. 16 HUH holds divalent metal ions to form nicks in the DNA strand in EN activity, whereas Tyr residues form a transient covalent bond with the cleaved DNA strand to generate phospho-tyrosine for DNA strand transfer. 16 The EN activity is abolished by mutation of either 2 His or 2 Tyr residues. 17 By aligning the 5 RHZs with reference REP domains, we found that the 5 RHZs conserved the 2 His and 2 Tyr residues (red asterisks in Figure 4B and Table 3). Taken together, our results suggest that the 5 rhzs encode both potentially functional HEL and EN domains (Table 3 and Supplementary Table 6). These 5 rhzs are located only on chromosome II (Figure 4C).
TDPGs in MPs in C. elegans
Our RepeatMasker analysis did not identify MP elements. Dfam_3.1 library contains 9 copies of Maverick and 20 copies of Polinton, excluding the absence of these elements from the repeat library as a potential cause. One possible cause is that MP is more than 10 kbp in length, longer than other TEs identified here; optimizing the parameters of RepeatMasker’s algorithm may be necessary for efficient identification. Notably, MPs located on chromosomes I, II, III, IV, and X of the C. elegans genome assembly (PRJNA907379) have previously been reported.21,22,82 Using the representative AL110478.1 sequence of MP in C. elegans, 82 we searched for MP copies in the VC2010 assembly by using the NCBI nucleotide BLAST program (https://blast.ncbi.nlm.nih.gov/). We identified short homologous regions >100 bps that were densely scattered on 3 distinct regions on chromosome I (MP Ia, MP Ib, and MP Ic) and 2 regions on chromosome X (MP Xa and MP Xb) (Figure 5A and B). Thus, the reference MP copy used in our homology search was aligned discontinuously in these putative MP copies in the VC2010 genome assembly (Figure 5B), and these MP copies were located on different chromosomes from previous reports.

Maverick/Polintons (MPs) in the Caenorhabditis elegans genome. (A) Genomic positions of homologous regions with AL110478.1. Five rectangular regions are as follows: in chromosome I: MP Ia, 3543 bp region from 0.4521550 × 107 to 0.4525093 × 107 bps; MP Ib, 17 276 bp region from 1.0486778 × 107 bps to 1.0504054 × 107 bps, and MP Ic, 43 817 bp region from 1.3280931 × 107 bps to 1.3324748 × 107 bps; and in chromosome X: MP Xa, 11 758 bp region from 0.2000999 × 107 bps to 0.2012757 × 107 bps, and MP Xb, 183 668 bp region from 1.7462913 × 107 bps to 1.7646581 × 107 bps. (B) Scattered distribution of homologous DNA regions with AL110478.1 in each of 5 MP copies. Red dots indicate homologous regions > 500 bps. Blue dots indicate homologous regions of <500 bps and >100 bps. (C) Alignment of catalytic core domains of INZ_MP-1 and INZ_MP-2 with reference Integrases (INs). Abbreviations: ASV: IN in avian sarcoma virus [1ASU_A], HERVK: Pol protein in human endogenous retrovirus K [CAA76885], MMLV: p46 IN Moloney murine leukemia virus (MoMLV) [NP_955592.1], HIV: IN in HIV-1 [1BIZ_A], Gypsy_Dm: IN, Gypsy endogenous retrovirus in Drosophila melanogaster [CAB69645], TEDV_Tm: ORFB in TED virus in Trichoplusia ni [YP_009507248], TP1731: Pol polyprotein in transposon_1731 in D. melanogaster [S00954], RVRP_At: retrovirus-related like Polyprotein in Arabidopsis thaliana [CAB78488_1], Copia_Dm: Gag-Int-Pol protein in COPIA in D. melanogaster [P04146], Ty-3_Sc: Gag-Pol polyprotein in Ty3-G in Saccharomyces cerevisiae [GFP69998.1]. (D) Alignment of catalytic core domains of INZ_MP-3 with reference Transposases (TPs). Amino acid sequences of Mariner-10_HM, Mariner-3_AN, Pogo_SM, Mariner-1_SP, CIRT2_CA, Mariner-1_AF, MAR1_TV, and Tc1-1_AG as examples of Tc1/mariner class TPs were arbitrarily obtained from Yuan and Wessler. 65 Asterisks indicate conserved amino acids in catalytic core.
The VC2010 genome assembly provides substantial advantages over its predecessors in both precision and completeness, but at the current stage, any assembly is imperfect. 39 Compared with the N2 genome assembly, the VC2010 assembly contains short insertions, deletions, and duplications ranging from tens to thousands of base pairs, which are distributed in all chromosomes. 39 These differences could be due to polymorphism in the VC2010 strain or could be due to errors arising during sequencing and/or assembly of the N2 genome assembly. In the comparison between recent de novo assemblies of the N2 and Hawaiian genomes, indels with about 50 bases are widely detected across the genomes, 83 suggesting that such small indels may be inserted at a relatively rapid rate in evolution. We interpret that the discontinuity within the MP copies (Figure 5A and B) reflects the actual sequence present in the VC2010 strain. On the other hand, as mentioned in previous reports, 39 artifactual large structural variations in VC2010 could still remain even after careful correction in the VC2010 assembly. At this point, we remain agnostic as to whether the different chromosomal locations of the MP copies in VC2010 are actually present in the strain or reflect errors in the assembly process to obtain the VC2010 assembly.
To identify TDPGs encoded in these MP loci, ie, DNA polymerase B (DNA POLB) and IN, we applied SNAP and DIAMOND to the 5 MP copies. Two DNA POLB-related genes (polB_MP) encoding 378 and 93 amino acids were located in MP Ia and MP X, respectively (Supplementary Table 7). Two IN genes (inz_MP) were located in MP Ib and MP Ic (Supplementary Table 8). Interestingly, a gene encoding the helix-turn-helix 48 (HTH48) domain-containing protein, which is conserved in some Transposases (TPs), is located in MP Xa (Supplementary Tables 9 and 10). Because TPs are functionally and evolutionarily related to INs,84,85 we considered these 3 IN-related genes to be inz_MPs.
DNA POLB has 5 conserved motifs from Motifs I to V. 86 By aligning PolB_MP-1 (at MP Ib) and PolB_MP-2 (at MP Xa) with the reference DNA POLBs, we found that PolB_MP-1 lacked most of the N-terminal motifs from I to III and only had YnDTD conserved in Motif IV. In addition, PolB_MP-2 only had conservation of a short N-terminus fragment and did not exhibit conservation of Motifs I to V. Therefore, we considered that these PolB_MPs likely do not encode functional proteins. Next, by aligning INZ_MP-1 (at MP Ib), INZ_MP-2 (at MP Ic), and INZ_MP-3 (MP Xa) with reference INs, we found that INZ_MP-1 and INZ_MP-2 encoded glutamic acid triads that align with the reference DDE triad (black asterisks in Figure 5C). Based on this potential functional conservation, we do not reject the possibility that inz_MP-1 and inz_MP-2 encode functional genes. INZ_MP-3 did not align with the reference DDE triads, whereas INZ_MP-3 aligned with the DDD/E triad in the Tc1 family TPs (red asterisks in Figure 5C). We thus also considered inz_MP-3 to be a potentially functional gene.
The IN encoded in a MP copy has been identified as a cellular IN (c-Integrase) that is homologous to the retrotransposon IN. 22 In addition, the IN in MP is highly homologous to the TP encoded in ginger DNA transposon. 87 Maverick/Polintons has been proposed to be involved in gene transfer between eukaryotic DNA mobile elements (dsDNA viruses, adenoviruses, small ssDNA viruses, Mavirus-like virophages, icosahedral viruses).88-90 Considering that inz_MP-3 at MP Xa encoded an HTH48 domain, and that the DDD motif was homologous to the Tc family of DNA TPs, we note the possibility of evolutionary interactions between the Tc family of DNA transposons and MP in C. elegans.
TDPGs in DIRS, PLE, and Crypton elements in C. elegans
Our RepeatMasker analysis did not identify copies of DIRS or Crypton. A previous homology-based search of 34 nematode species to identify Tyr-REC genes that mediate transposition of DIRS and Crypton identified an incomplete cDNA for a Tyr-REC gene in chromosome II of C. elegans. 91 We applied SNAP to a genomic region of this incomplete cDNA with 20 kbp 5′ flanking and 20 kbp 3′ flanking genomic regions in VC2010, but did not find any complete ORF. From this result and previous studies, we conclude that C. elegans may not have a functional Tyr-REC gene. Finally, consistent with a previous report, 92 our analysis on VC2010 did not identify any PLE copies.
Discussion
Extensive bioinformatic studies on TEs have led to the identification of novel TEs, the inferred mechanisms of transposition from enzymes coded on TEs, and through phylogenetics analysis, the clarification of the evolutionary processes of TEs and the genome.6,7,26 Despite such studies, the physiological functions of TEs, such as their roles in development, and aging, as well as evolution, are largely unverified experimentally. In this report, we searched for TDPGs in all the representative TE orders in the latest genome assembly of C. elegans (VC2010). According to our RepeatMasker analysis, more than 50 000 TE loci exist in the latest C. elegans genome assembly (Figure 6A and Supplementary Table 1). These loci encode 428 complete ORFs, 66.8% of which (285 genes) are TDPGs. Among them, we identified 142 potentially functional genes, including 8 rtz_LTRs, 7 inz_LTRs, 28 rtz_LINEs, 94 tpzs, and 5 rhzs (Figure 6B). In addition, our manual analysis identified 3 potentially functional inz_MPs (Figure 6B). In total, we identified 145 potentially functional TDPGs.
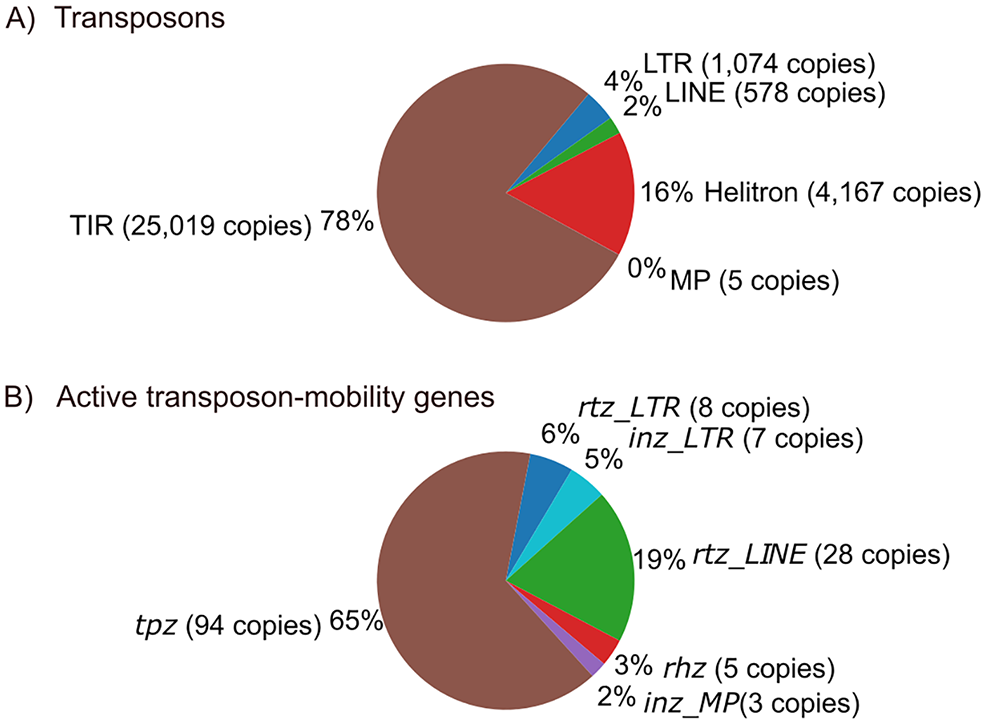
Summary of potentially functional TDPGs in the Caenorhabditis elegans genome. (A) The percentage of copy numbers of LTR element (blue), LINE (green), Helitron (red), MP (purple), and TIR element (brown) in the total copy number of TEs in the C. elegans genome. (B) The percentage of potentially functional TDPGs; 8 rtz_LTR (blue) and 7 inz_LTR (light blue), 28 rtz_LINE (green), 5 rhz (red), 3 inz_MP (purple), and 94 tpz (brown) in total 145 potentially functional TDPGs identified in this study.
The preponderance of total TE loci relative to those retaining potential for mobility is well noted but has rarely been quantified. The best studied example is the retrotransposon L1, a member of the well-studied LINE order. The human genome has more than 500 000 loci corresponding to L1, accounting for 17% of the human genome. 93 However, bioinformatic analysis has identified only 146 copies of full-length L1 in the human genome, and only 107 of these L1 copies have conserved intact RT genes. 94 In vitro experimental tests of the mobility of L1 copies showed that fewer than 100 copies of L1 were active, with 6 L1 copies accounting for most of the activity of L1 in the human genome. 95 Similarly, cancer cell genome analysis and genome comparison in human population showed that limited number of L1 loci comprise the bulk of mobile activity of L1.96-99 These results indicate that a limited number of L1 copies encode functional TDPGs, which is consistent with our finding of the stark contrast between the number of potentially functional TDPGs and the total number of TEs.
Critical amino acids at the catalytic cores of TDPGs have been studied in LTR elements,27-29 LINEs,30-32 and TIR elements 33 in C. elegans. We compared the 145 potentially functional TDPGs identified in this study to those identified previously. In previous studies of LTR elements, researchers identified 2427 or 1028 full-length copies among 124 or 62 LTR element copies, respectively, in C. elegans. Another study reported 17 rtz_LTRs with conserved Asp residues in Motifs A and C, and 15 inz_LTRs with a conserved DDE triad. 29 We identified 8 potentially functional rtz_LTR and 7 potentially functional inz_LTR genes. Thus, we found fewer potentially functional rtz_LTRs and inz_LTRs in this study than were identified by previous studies.27-29 Differences in the genome assembly used in Ganko et al 27 and Bowen and McDonald, 29 as discussed in Yoshimura et al, 39 might contribute to this discrepancy. On the other hand, using the same VC2010 assembly as us, Kanzaki et al 28 identified 10 LTR elements; we identified nearly the same number but slightly fewer TDPGs. A detailed analysis of the coding sequence of the catalytic core of enzymes in the 10 full-length LTR elements 28 may provide consilience with the number of potentially functional LTR element genes identified here. Notably, TDPGs in the most-studied C. elegans LTR element, Cer1 on chromosome III,27,100 were found on our list of potentially functional genes (i.e. rtz_LTR-5 and inz_LTR-5; Figure 1C). Cer1 is both biologically active and mobile101-103 in recent evolutionary history, based on a comparison of natural isolates of C. elegans. 104 For LINE retrotransposons, among more than 1000 copies of LINE, 30 6 copies of the RTE LINE suborder 31 and 17 copies of the T1/CR1 LINE suborder 32 encode the rtz_LINE that conserves the Asp residues in Motifs A and C. We identified a similar number of LINE copies (618 copies; Supplementary Table 1). Among them, 28 rtz_LINEs preserved potentially functional RT domains, which is more than total number of potentially functional rtz_LINEs reported previously. For TIR elements, a previous report showed that 61 tpzs had the conserved DDD/E motifs in 127 copies of Tc/mariner family TIR element. 33 We identified 94 potentially functional tpzs in 189 copies of the Tc/mariner family TIR element, which is a larger number than was reported previously. For Helitron DNA transposon, we found that 1.65% of the C. elegans genome was occupied with Helitron copies (Supplementary Table 1), similar to previous reports (~2%). 18 There was no further amino acid sequence analysis about rhzs.
In addition, we investigated the latest version of Wormbase (WS292). For Tc1/mariner, among its 636 copies, 129 tpzs were found, with 112 being potentially functional tpzs. Regarding LINE, among its 62 copies, 9 out of 10 potentially functional rtz_LINEs were found. For LTR element, among its 15 copies with 4 ORFs registered, none showed potentially functional rtz_LTR and inz_LTR. In the case of MP, among its 12 copies, 3 polB_MPs (C33E10.6, Y106G6G.5, Y26D4A.9) were registered, but inz_MP was not found. Among these PolB_MP, only Y26D4A.9 preserved motif IV out of 5 conserved motifs, and thereby these pol_MPs were not considered potentially functional genes. Likely due to discrepancies in the genome sequences, corresponding ORFs for these 3 polBs were not found in the VC2010 genome assembly. Helitron, DIRS, Crypton, and PLE were not registered. Taken together, our catalog of potentially functional TDPGs includes an equivalent or, for some TEs, greater number of genes compared with the number identified in previous reports, and reflects various lines of available biological and bioinformatic evidence, supporting the validity of our annotation.
In human and mouse genomes, L1 is the most abundant among TE classes, with 145 copies in human and 2811 copies in Mus musculus being conserved at full length.94,105,106 In D. melanogaster, Saccharomyces cerevisiae, and Arabidopsis thaliana, 107 LTR element is the most abundant class, with 325 copies in D. melanogaster 108 and 51 copies in S. cerevisiae 109 being conserved at full length. Long terminal repeat elements are only the TE encoded in Schizosaccharomyces pombe, with 13 copies being conserved at full length. 110 DNA transposons are the most abundant in Danio rerio (zebrafish), with 2.3 million copies, but it is unknown how many of these copies are active. 111 Similarly, it is unknown how many of the 286 LTR elements in A. thaliana are active. 107 In our study, TIR elements were the most abundant transposon class (20 852 copies; Supplementary Table 1) in C. elegans, encoding 94 copies of potentially functional tpz. Our identification of 145 potentially functional TDPGs indicates that C. elegans encodes the fewest number of potentially functional TDPGs among conventional metazoan model organisms. We propose that C. elegans can be a useful model metazoan to study the conserved physiological, pathological, and evolutionary roles of transposon mobility.
Conclusions
Our TDPG catalog, which contains 145 potentially functional TDPGs encoded in LTR elements, LINEs, TIR elements, Helitrons, and Mavericks/Polintons, serves as a source of information for conducting RNA interference (RNAi) experiments to systematically manipulate the mobility of both autonomous and non-autonomous TEs. By designing primer sets to specifically amplify certain genome regions encoding a TDPG, RNAi experiments can simultaneously manipulate the mobility of TEs that rely on the target TDPGs, other TEs that rely on homologous copies of the TDPG, and non-autonomous TEs that depend on these TDPGs. RNAi targeting a TDPG in a homologous gene cluster of rtz_LINE and tpz (Supplementary Figures 1 and 2) can more efficiently and simultaneously affect the mobility of multiple TEs. Our TDPG catalog could potentially promote the study of the physiological functions of TE mobility.
Limitation
Our gene catalog provides substantial advantages over its predecessors, such as Wormbase for conducting experimental studies but is imperfect. The TDPGs in our catalog were curated based on amino acid sequences within the catalytic cores; some of these genes might be truncated in other regions, such as the N- or C-terminus or within inter-functional domains. The TDPGs in our catalog might lack DNA elements necessary for proper gene transcription. Due to mutations in regions responsible for translational regulation, 112 it is possible that genes may not produce functional transcripts or proteins. In addition, our current TDPG search scheme does not detect genes located in the genome regions that RepeatMasker did not recognize as TEs. Thus, the possibility remains that there are still unidentified TDPGs. To address these shortcomings, it will be necessary to combine multiple algorithms for TE and gene identification across different versions of wild-type genome assemblies, based on continuously updated reference libraries for TEs, and to take the union of the TDPGs identified through these bioinformatics searches. In addition, integrating in vivo transcript information obtained from Expressed Sequence tags and RNA-seq data could further refine the gene catalog. Experimental evolution and re-sequencing organisms after targeting the current set of TDPGs would provide an orthogonal approach to evaluate whether this catalog is exhaustive. These expanded analyses could provide a more rigorous basis for experimental manipulation of TE mobility in vivo.
Supplemental Material
sj-tiff-1-bbi-10.1177_11779322241304668 – Supplemental material for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans
Supplemental material, sj-tiff-1-bbi-10.1177_11779322241304668 for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans by Yukinobu Arata, Peter Jurica, Nicholas Parrish and Yasushi Sako in Bioinformatics and Biology Insights
Supplemental Material
sj-tiff-2-bbi-10.1177_11779322241304668 – Supplemental material for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans
Supplemental material, sj-tiff-2-bbi-10.1177_11779322241304668 for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans by Yukinobu Arata, Peter Jurica, Nicholas Parrish and Yasushi Sako in Bioinformatics and Biology Insights
Supplemental Material
sj-xls-3-bbi-10.1177_11779322241304668 – Supplemental material for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans
Supplemental material, sj-xls-3-bbi-10.1177_11779322241304668 for Bioinformatic Annotation of Transposon DNA Processing Genes on the Long-Read Genome Assembly of Caenorhabditis elegans by Yukinobu Arata, Peter Jurica, Nicholas Parrish and Yasushi Sako in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
We thank Dr Damien Farrell at University College Dublin for the sequence alignment viewer. We also would like to thank Thomas J. LaRocca at Colorado State University for his helpful comments and suggestions on manuscript. Finally, we extend our gratitude to WormBase (![]() ) for a valuable help for our analysis.
) for a valuable help for our analysis.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by JSPS KAKENHI grant number 20K20321 (Grant-in-Aid for Challenging Research (Exploratory)) (Y.A.).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
Y.A. and N.P. conceptualized the study. Y.A. and P.J. performed the data analysis. Y.S. provided supervision, contributed to the interpretation of the results, and critically reviewed the manuscript. Y.A. drafted the manuscript.
Supplemental material
Supplemental materials for this article are available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.