
Abstract
Estrogen receptor alpha (ERα) plays a critical role in breast cancer (BC) progression, with endocrine therapy being a key treatment for ERα + BC. However, resistance often arises due to somatic mutations in the ERα ligand-binding domain (LBD). Lasofoxifene, a third-generation selective estrogen receptor modulator, has shown promise against Y537S and D538G mutations. However, the emergence of a novel F404 mutation in patients with pre-existing LBD mutations raises concerns about its impact on lasofoxifene efficacy. This study investigates the impact of the dual Y537S and F404V mutations on lasofoxifene’s efficacy. Using molecular dynamics simulations and molecular mechanics/Poisson-Boltzmann surface area (MM-PBSA) free energy calculations, we found that the dual mutation reduces lasofoxifene binding affinity and binding free energy, disrupts crucial protein-ligand interactions, and induces significant conformational changes in the ligand-binding pocket. These alterations are likely due to the loss of the pi-pi stacking interaction in the F404V mutation. These findings suggest a potential reduction in lasofoxifene efficacy due to the dual mutation. Further experimental validation is required to confirm these results and fully understand the impact of dual mutations on lasofoxifene’s effectiveness in ERα + metastatic BC.
Introduction
Estrogen receptor alpha (ERα) is a ligand-activated nuclear hormone receptor, expressed in roughly 70% of breast cancer (BC) cases, 1 plays a crucial role in BC development and progression. Targeting ERα signaling with endocrine therapy (ET) remains a cornerstone in treating early and metastatic ERα + BC. 1 This therapeutic strategy revolves around 3 main strategies: (1) using aromatase inhibitors (AIs) to systematically reduce the endogenous estrogen levels and deprive the receptor of its ligand, (2) using selective estrogen receptor modulators (SERMs), such as tamoxifen, raloxifen, and toremifen, to inhibit the binding of estrogen to its receptor, 2 and (3) using selective estrogen receptor down-regulators (SERDs), such as fulvestrant and AZD9496, to degrade ERα.3,4
Despite initial success, resistance emerges in a significant portion of metastatic breast cancer (MBC) patients receiving endocrine therapy. Somatic gain-of-function mutations in the ERα ligand-binding domain (LBD), occurring at a frequency of 30% to 40%, are the major contributor to this resistance.5-8 These recurrent mutations arise under the selective pressure of ET treatments, particularly, in patients treated with AIs.5-8Among these, the Y537S/C/N mutations are most frequent, with Y537S exhibiting the highest resistance to endocrine therapy.6,9 Studies suggest that these mutations activate the receptor even in the absence of estrogen, promoting co-activator recruitment and resistance to traditional therapies.10-12
Lasofoxifene, a third-generation SERM initially developed for postmenopausal vaginal atrophy and osteoporosis, recently showed promise in overcoming endocrine resistance in ERα + MBC models harboring Y537S or D538G mutations. 13 Preclinical research indicates lasofoxifene effectively suppresses tumor growth and metastases compared to fulvestrant in models with Y537S mutations, by disrupting their constitutive agonist conformation. 13 Furthermore, phase 2 clinical trials demonstrate encouraging antitumor activity and tolerability for lasofoxifene as monotherapy in patients with ET-resistant, ERα + mutated MBC.13-15 These findings suggest lasofoxifene’s potential as an efficient treatment for such patients class.
However, a recent study identified a novel LBD mutation, F404, in ~4% of patients who progressed on fulvestrant. 16 This mutation, often acquired in patients with pre-existing LBD mutations like Y537S or D538G, disrupts the ligand-receptor interaction and cause drug-specific resistance particularly to the widely used SERD, fulvestrant. 16
Building upon this context, our study aims to investigate the impact of the dual Y537S and F404V (Acquired in ~1%) ERα mutations on lasofoxifene’s efficacy, specifically, exploring whether lasofoxifene retain its efficacy in overcoming resistance mechanisms when both Y537S and F404V mutations are present. To achieve this, we conducted a comparative molecular dynamics study, comparing the mutated Y537S in complex with lasofoxifene to the double Y537S + F404 mutant model in complex with lasofoxifene.
Materials and Methods
Structures preparation
The crystal structure of mutated Y537S ERα in complex with lasofoxifene (PDB id: 6VGH) was used as reference model. The missing residues between R335–S341, S417–G420, F461–T465, and Y526–N532 were modeled using modeler implemented in Chimera version 1.4. 17 The Y537S + F404V mutant ERα model was built by making point mutation F404V on the 6VGH model with the Rotamers tool of UCSF Chimera. 18 Then, the mutant model was relaxed by 1000 steps of SD followed by 1000 steps of CG minimizations keeping all atoms far by more than 5Å from the mutated residue fixed.
Binding affinity of the Lasofoxifene ERα docked complex
To investigate the impact of the F404V mutation, AutoDock Vina score_only method was used to predict changes in ligand-receptor affinity. 19 In addition, two-dimensional (2D) diagrams were used to analyze the interaction mode and assess the mutation effect on ligand-receptor interactions.
Molecular dynamics simulation
The MD simulations were conducted for 100 ns with a recording interval of 50 ps using the academic Desmond software package. 20 In system builder, the OPLS3e force field was selected and TIP3P was used as a solvent model with a 10Å padding orthorhombic box, then the system charge was neutralized by adding 0.15M of sodium (Na+) and chloride ions (Cl−). Following this, the generated model systems underwent energy minimization and equilibration via the NPT ensemble at a constant temperature of 300 K and pressure of 1.01325 bar. All other Desmond parameters were maintained at their default values. Subsequently, simulation trajectories were analyzed using the simulation interaction diagram included in Desmond. 20
MM/PBSA free energy calculations and residue decomposition
The MM/PBSA free energy was calculated using the gmx_MMPBSA tool, which requires .top, and .trr files as input. To obtain these, we first converted the Desmond Composite Model System files (.cms) using InterMol software (https://github.com/shirtsgroup/InterMol) to generate “.gro” and “.top” files. In addition, the Desmond trajectory was imported into VMD and saved under “.trr” format. Once all required input files were prepared, the MM-PBSA was calculated using the following command:

- i mmpbsa.in: Specifies the input file containing MM/PBSA parameters and settings.
- cs complex.pdb: Specifies the coordinate file in .pdb format.
- ci index.ndx: Specifies the index file that defines the groups of atoms for the calculations.
- ct trajectory.trr: Specifies the trajectory file in .trr format.
- cp topol.top: Specifies the topology file in .top format.
- o MMPBSA.dat: Specifies the output file for the main MM/PBSA calculation results in .dat format.
- eo MMPBSA.csv: Specifies the output file for the main MM/PBSA calculation results in .csv format.
- do MMPBSA_decomposition.dat: Specifies the output file for the decomposition analysis results in .dat format, showing the contribution of individual residues or groups of residues.
- deo MMPBSA_decomposition.csv: Specifies the output file for the decomposition analysis results in .csv format.
Results
Y537S + F404V double mutation reduces lasofoxifene binding affinity
In this study, we investigated the efficacy of lasofoxifene against the double mutation Y537S + F404V in ERα compared to its effectiveness against the single mutation Y537S. To achieve this, we first introduced the F404V mutation into the Y537S-mutated model to create a double-mutant ERα model (Y537S + F404V). Subsequently, the binding affinity scores were recalculated using the score_only function implemented in AutoDock Vina to depict the ligand-receptor binding affinity changes. 21
The rescoring results showed that introducing the F404V mutation in the Y537S-mutated model decreased the binding affinity score by 0.90 kcal/mol (from −10.40 to −9.50 kcal/mol). This decrement suggests a potential weakening of the interaction in the double mutation (Y537S + F404V) model in comparison to the single mutation (Y537S) model (Figure 1). To understand how the F404V mutation affects the binding affinity, we compared the interaction modes between ERα and lasofoxifene in the single-mutant and the double-mutant models (Table 1). This comparison revealed that all hydrophobic (Ala350, Leu354, Leu384, Ile424, Leu525 Leu525 and Leu536), hydrogen bonds (Glu353, Asp351, and Arg394), water bridge (Arg394), and salt bridge (Asp351) interactions were conserved, maintaining the same distances between the single mutant and the double mutant, except for the perpendicular pi-pi stacking interaction with amino acid 404, which was notably absent in the double-mutant model (Table 1). This discrepancy stems from the substitution of phenylalanine with valine at position 404, resulting in the loss of the aromatic ring necessary for pi-pi stacking interaction with a corresponding aromatic ring within lasofoxifene, which may explain the observed decrease in binding affinity (Figure 1).
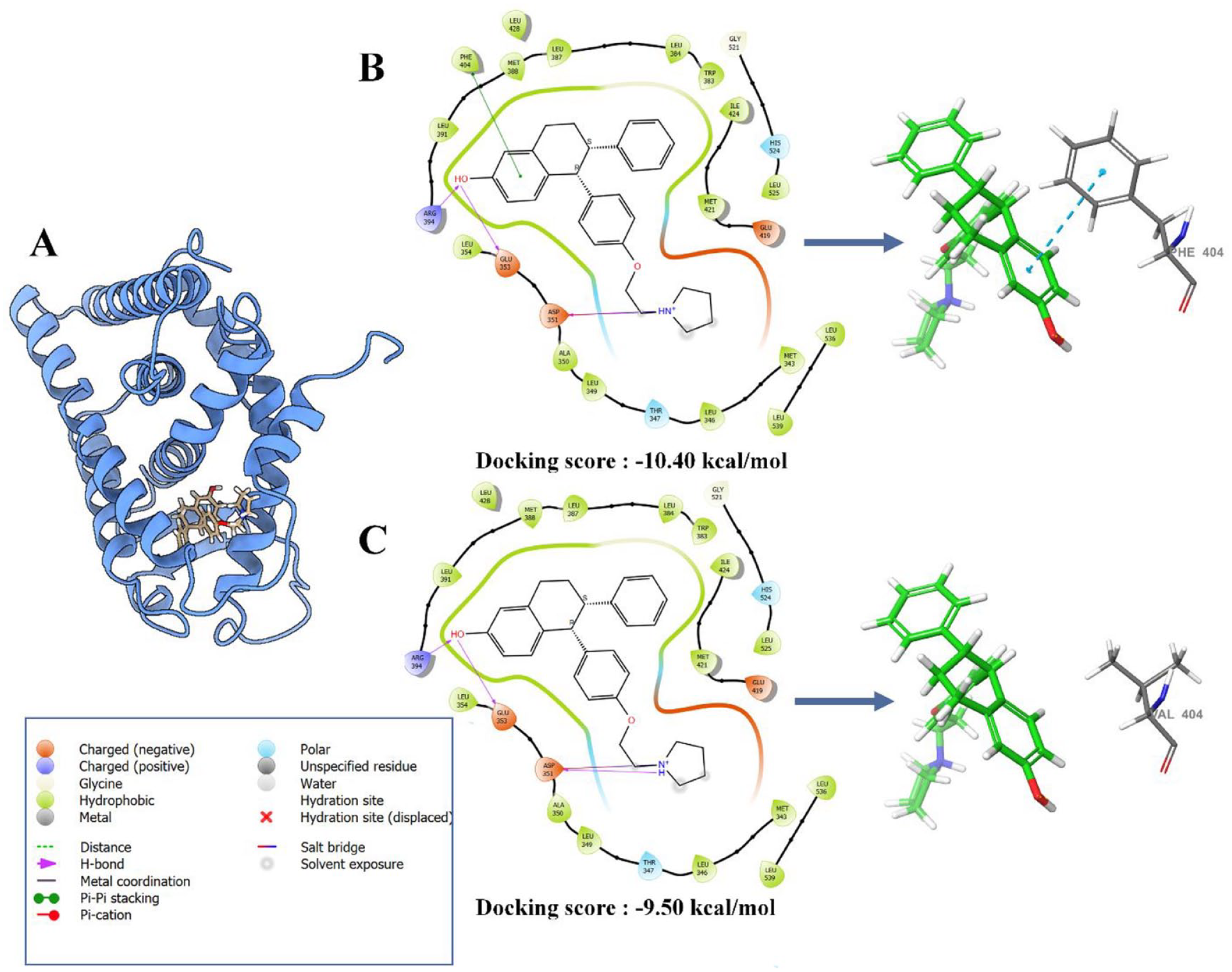
Docking poses (A), 2-D diagram interaction and binding affinity of lasofoxifene in complex with single (B) and double mutant (C) model of ERα.
Summary of the main interatomic interactions between ERα and lasofoxifene in single (Y537S) and the double-mutant (Y537S + F404V) models.

(+) indicates the presence of interaction and (–) indicates the absence of interaction.
Y537S + F404V double mutation alters conformational dynamics and stability
To assess whether the double mutation disrupts the conformational dynamics of the ERα and its binding affinity to lasofoxifene, we conducted a comparative molecular dynamics study, comparing the behavior of the single-mutant model (Y537S) to that of the double-mutant model (Y537S + F404V) of ERα in complex with lasofoxifene. The molecular dynamics results were analyzed using various parameters such as (1) protein root mean square deviation (P-RMSD) that measures the conformational changes of given complex over time and describes whether the simulation is in equilibrium (Figure 2A), (2) protein root mean square fluctuation (P-RMSF) that characterizes local changes along the protein chain (Figure 2B), and (3) protein-ligand contact analysis that presents the fraction of the active residues implicated in the ligand interaction (Figure 3). 22

Protein RMSD (A), ligand pocket RMSD (B), and protein RMSF (C) of single (Y537S) and double-ERα-mutant (Y537S + F404V) models in complex with lasofoxifene.

Monitoring of protein-ligand contacts of bound lasofoxifene to single (A) and double-ERα-mutant (B) models throughout simulation. The left histograms showing interaction fraction with active amino acid residues. The x axis presents the residues involved in the interactions, and y axis presents the normalized value of the temporal length of the interactions during the simulation. The stacked bar charts are normalized over the course of the trajectory: for example, a value of 0.7 suggests that 70% of the simulation time the specific interaction is maintained. Values over 1 indicates that the protein residue could make multiple contacts of the same subtype with the ligand. The middle schematic representation (2D simulation interaction diagram) of ligands indicating percentage of interactions with the protein residues. The right plot indicating timeline of protein-ligand contacts during the simulation. Some residues made more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot.
The RMSD analysis revealed distinct dynamics between the single-mutant model and the double-mutant model of the ERα during the MD simulation (Figure 2A). Indeed, the single mutant model reached equilibrium after 30 ns of simulation, maintaining stability with minor fluctuations until the end of the simulation with an average RMSD value of 3.12 Å. In contrast, the double-mutant model exhibited higher fluctuations, particularly between 15 and 60 ns, followed by notable stability between 60 and 80 ns, before undergoing further destabilization, resulting in an overall RMSD value of 3.53 Å. This suggests that introduction of the double mutation leads to structural alterations that affect the global receptor stability, leading to increased structural fluctuations over time.
Subsequently, we focused on the RMSD of the ligand-binding pocket, encompassing residues within 5 Å of lasofoxifene to assess ligand-binding stability and detect any conformational discrepancies resulting from the double mutation (Figure 2B). The results indicated a higher RMSD value for the double mutant (average of 5.21 Å), compared to the single mutant (average of 4.79 Å), indicating significant conformational changes within the ligand binding pocket induced by the double mutation.
Following the RMSD analysis, RMSF analysis was conducted to further investigate the local flexibility of both models throughout the simulations. The RMSF analysis revealed some significant differences between the 2 models, particularly in the loop connecting helices H11 and H12 (between 526 and 530), where the RMSF value was notably higher (exceeding 6Å) in the double-mutant model compared to the single-mutant counterpart (4.1 Å). Notably, the region between residues 368 and 374 and the loop connecting helices H11 and H12 constitutes part of the ligand-binding pocket, which can explain the high RMSD value observed in this pocket. In addition, regions 456 to 465, 415 to 418, and 368 to 374 exhibited fluctuations higher by 2 Å, 1.5 Å, and 1 Å, respectively, compared to the single-mutant model. For the other regions of the protein, the difference in fluctuation was similar or less than 1 Å (Figure 2C).
Y537S + F404V double mutation disrupts protein-ligand interactions
The conformational changes observed around the ligand-binding pocket following the introduction of the F404V mutation to the single-mutant model, suggest a potential alteration in accommodation and recognition of lasofoxifene within the ligand-binding pocket, by disrupting the specific interactions between the protein and the ligand, leading to a change in their binding mode. To better understand these potential consequences, the protein-ligand interactions were monitored and compared systematically throughout the 100 ns of the MD simulation (Figure 3).
The obtained results revealed significant differences between the double-mutant and single-mutant models. Particularly striking was the absence of the hydrophobic interaction, attributable to pi-pi stacking, between residue 404 and lasofoxifene in the double-mutant model, while this interaction is present in the single-mutant model with an occupancy of 57% of the simulation duration. In addition, another water bridge interaction was observed with residue 404 in the single-mutant model, with an occupancy of 22%, whereas in the double-mutant model, this interaction did not exceed 18% of the simulation. This short water bridge interaction cannot compensate for the missing pi-pi stacking interaction.
Beyond residue 404, several differences in the occupancy of specific interactions were observed between single- and double-mutant models. Specifically, at residues 350 and 353 in helix 3, residues 383, 384, and 387 in helix 5/6 and residue 530 in the loop connecting H11–H12. First, residue 383 in the single mutant engaged in both H-bond (occupancy of 15%) and hydrophobic interactions (occupancy of 13%), whereas in the double mutant, only a hydrophobic interaction was detected, with an occupancy of less than 10%. Similarly, residue 384 in the single mutant exhibited a hydrophobic interaction with an occupancy of 27%, contrasting with a reduced occupancy of 8% in the double mutant. Moreover, residue 387 in the single mutant formed 2 interactions, a hydrophobic interaction (occupancy of 22%) and a water bridge interaction (occupancy of 54%), whereas in the double mutant, the hydrophobic interaction occupancy decreased to 12%, and the water bridge interaction occupancy decreased to 38%. Furthermore, residue 530 formed a water bridge interaction in the single mutant with an occupancy of 26%, which was nearly absent in the double mutant, with an occupancy of 4%. This absence of interaction in residue 530, located in the loop connecting helices 11 and 12, may contribute to the higher flexibility observed in this loop. Finally, residue 353 in the single mutant engaged in a water bridge interaction with a high occupancy of 61%, whereas in the double mutant, residue 353 formed 2 interactions; one H-bond with an occupancy of 22% and 2 water bridge interactions with occupancies of 45% and 43%, respectively.
Overall, the monitoring of ERα-lasofoxifene interactions throughout the MD simulations suggests that conformational changes around the binding pocket result in significant variations in interactions within the double mutant model.
Y537S + F404V double mutation decreases lasofoxifene binding free energy
To examine whether the changes in the binding mode can lead to differences in the calculated binding free energies between both systems, we performed an MM-PBSA analysis. As a result, the MM-PBSA analysis revealed a notable decrease in the binding affinity of lasofoxifene in double mutant (−31.88 kcal/mol) compared to the single mutant (−35.29 kcal/mol) (Figure 4A). Further examination through detailed free-energy component analysis revealed that ΔGgas was predominantly influenced in the double mutant. Specifically, there was a substantial decrease in ∆Evdw (−4.91 kcal/mol) and a minor decrease in ∆Eelec (−0.61 kcal/mol), leading to an overall reduction in ΔGgas (−5.53 kcal/mol) (Figure 4).
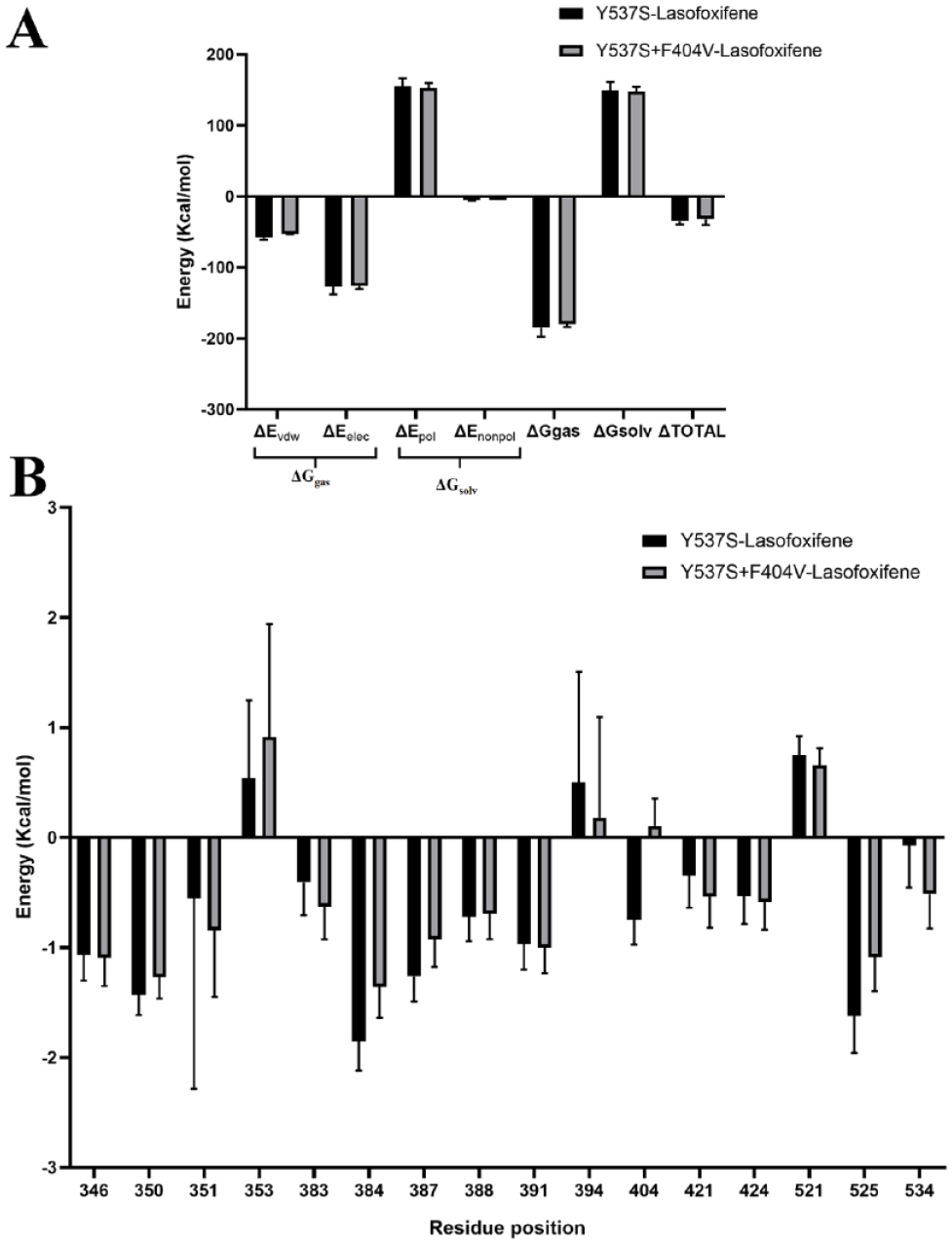
MM/PBSA binding free energy (A) and per residue free energy decomposition (B) for lasofoxifene in complex with single (Y537S) and double mutant (Y537S + F404V) ERα.
To gain deeper insights, a per-residue free energy decomposition analysis was performed to investigate the contributions of individual amino acid residues surrounding the binding site to the overall binding free energy (Figure 4B). The introduction of the F404V mutation led to less-favorable energy contributions from several critical residues, particularly at positions 350, 353, 384, 387, 404, and 525. This explains the observed decrease in the MM-PBSA binding free energy and alterations in the binding dynamics. Notably, residue 404, where the mutation occurs, exhibited a negative energy contribution (positive value) in the double mutant compared to the single mutant, with the energy changing from −0.74 to 0.12 kcal/mol (Figure 4B).
Conclusion
This study highlights the significant impact of the double Y537S + F404V mutation on the binding mode and affinity of lasofoxifene to ERα. The introduction of the F404V mutation led to a reduction in binding affinity by 0.90 kcal/mol and disrupted critical pi-pi stacking interactions. Molecular dynamics simulations revealed increased structural fluctuations and alterations in the ligand-binding pocket, resulting in diminished interaction stability. Per-residue free energy decomposition further identified specific residues contributing to the decreased binding affinity. These findings enhance our understanding of mutation-induced changes in ERα dynamics and binding, offering insights into the potential impact of this double mutation on the efficacy of lasofoxifene. However, further experimental validation is needed to confirm these results.
Footnotes
Acknowledgements
The authors thank Mohammed VI Center for Research and Innovation for providing resources and support.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
E.B. Conceptualization, Methodology, Software, Validation, Formal analysis, Investigation, Resources, Project administration, Visualization, Writing - Original Draft.
M.H. Software, Validation, Resources, Review & Editing.
Consent
This study did not involve human participants, so informed consent was not required.
Ethics
This study did not involve human or animal subjects, and therefore, ethical approval was not necessary.