
Abstract
Background:
The ability to predict and comprehend molecular interactions offers significant insights into the biological functions of proteins. The interaction between surface protein 47 of Plasmodium falciparum (Pfs47) and receptor of the protein 47 (P47Rec) has attracted increased attention due to their role in parasite evasion of the mosquito immune system and the concept of geographical coevolution between species. The aims of this study were as follows: to apply a bioinformatics approach to investigate the interaction between Pfs47 and P47Rec proteins and to identify the potential binding sites, protein orientations and receptor specificity sites concerning the geographical origins of the vectors and the parasite.
Methods:
Public sequences of the pfs47 and p47rec genes were downloaded and subsequently filtered to predict functional and structural annotations of the Pfs47-P47Rec complex. Phylogenetic analyses of both proteins were carried out. In addition, the p47Rec gene was subjected to sequencing and subsequent analysis in 2 distinct Anopheles species collected in Honduras.
Results:
The examination of motifs reveals a significant degree of conservation in pfs47, suggesting that Pfs47 might have undergone recent evolutionary development and adaptation. Structural models and docking analyses supported the theory of selectivity of Plasmodium falciparum strains towards their vectors in diverse geographical regions. A detailed description of the putative interaction between the Pfs47-P47Rec complex is shown.
Conclusions:
The study identifies coevolutionary patterns between P47Rec and Pfs47 related to the speciation and geographic dispersion of Anopheles species and Plasmodium falciparum, with Pfs47 evolving more recently than P47Rec. This suggests a link between the parasite’s adaptability and existing anopheline species across different regions. P47Rec likely has a cytoplasmic localization due to its lack of membrane attachment elements. However, these findings are based on simulations and require validation through methods like cryo-electron microscopy. A significant limitation is the scarcity of sequences in global databases, which restricts precise interaction modelling. Further research with diverse parasite isolates and anopheline species is recommended to enhance understanding of these proteins’ structure and interaction.
Background
Recent studies have provided an in-depth understanding of the relationship between the protein Pfs47 of Plasmodium falciparum, the main pathogen responsible for malaria in humans and the protein P47Rec present in the midgut cells of anopheline mosquitoes.1-3 The interaction between these proteins seems to substantially influence the parasite’s capacity to proliferate within the mosquito. 4 Pfs47 has been identified as the main variable mediating parasites’ evasion of the mosquito immune system.2,3,5,6 Consequently, specific strains of P falciparum have an enhanced ability to persist and proliferate within the anopheline mosquito species that have undergone geographical coevolution with the parasite. On the contrary, it has been shown that P falciparum strains that infect Anopheles mosquito species with a geographical origin different from that of the parasite are effectively eliminated and encapsulated by the vector’s immune system. 7 Pfs47 haplotypes demonstrate compatibility with their sympatric vectors, but haplotypes that are incompatible with the mosquito fail to survive. 8 This has been called ‘the lock and key theory’, which proposes a high specificity between the Pfs47 haplotypes of the parasite and those of P47Rec of the mosquito.9,10
Pfs47 is a member of the 6-cysteine (6-Cys) protein family. 11 The gene pfs47 lacks introns, and the protein has a signal peptide and a glycosylphosphatidylinositol anchor (GPI) sequence as an anchor. Pfs47 consists of 3 domains, with domains 1 and 3 exhibiting patterns consistent with the 6-Cys family. In contrast, the central domain is characterized by the presence of only 2 cysteine residues, 12 which seem to be evolving under positive or directional selection placing Pfs47 among the strongest signatures of natural selection and population structure in the P falciparum genome. 13
In a recent study, Molina-Cruz et al employed a comprehensive biochemical approach to identify and elucidate the characteristics of the Pfs47 receptor (P47Rec). Their findings revealed that this is a highly conserved protein consisting of 2 natterin-like domains present in the midgut cells of anopheline mosquitoes. 8 The p47rec gene consists of 4 exons encoding a polypeptide comprising 290 amino acids. P47Rec is characterized by the presence of 4 repeats of the DM9 domain. 14 This receptor is located mostly on the apical side of midgut epithelial cells immediately below microvilli. The interaction between P47Rec and the ookinete surface of the parasite is observed during the process of invasion. P47Rec appears to accumulate at the specific site of invasion, where it colocalizes with Pfs25, a protein found in P falciparum that is also present on the surface of the ookinete.8,15 The surface of the parasite establishes proximity with P47Rec, leading to the localized aggregation of the receptor when ookinetes invade the apical region of midgut epithelial cells.
This study aimed to investigate the interaction between Pfs47 and P47Rec proteins to gain insights into the immune evasion mechanisms employed by P falciparum to survive in the mosquito midgut. To achieve this, molecular docking analysis was utilized to identify interaction models, and structural predictions of the protein complex were developed. In addition, we employed a bioinformatics methodology to delineate the potential binding sites, protein orientations and receptor specificity sites concerning the geographical origins of the vectors and the parasite.
Methods
Sequencing of p47rec gene of Anopheles albimanus and Anopheles darlingi from Honduras
Several sets of primers were designed to amplify partial fragments of the p47rec gene in 2 species of Anopheles collected in Honduras: A albimanus and A darlingi. The mosquitoes were collected in 2021. A albimanus specimens were captured in the department of ‘Gracias a Dios’, and A darlingi were collected in the department of ‘Atlántida’. Polymerase chain reaction (PCR) reactions were carried out in a final volume of 50 µL using 25 µL of Taq Master Mix 2× (Promega Corp. Madison, WI, USA), 1 µL of each primer (10 µM) (Table 1), 2 µL of DNA and 21 µL of nuclease-free water. The cycling conditions were as follows: 1 cycle of 95°C for 5 min, 35 cycles of 95°C for 60 s, 55°C for 60 to 90 s (Table 1) and 72°C for 60 s, with 1 final cycle of 72°C for 5 min. Amplified products were visualized on 1% agarose gels with ethidium bromide. The products were sequenced on both sides. Sequences were trimmed, edited and assembled in Geneious Prime 2023.2 software. Two sequences of each species were deposited in GenBank. Sequences from mosquitoes captured in Honduras were included in the study since the sequences available in the databases correspond to specimens collected in Mexico and South America.
Sequences, amplicon size and annealing temperature of the primers used to amplify the Anopheles p47rec gene.

Pipeline
A pipeline to generate predictions at multiple levels of functional and structural annotations from nucleotide sequences of the genes that encode the proteins of the Pfs47-P47Rec complex was developed. The putative molecular interactions and evolutionary processes involved in the emergence and speciation of the 2 proteins of the complex were predicted (Figure 1). As an initial strategy, public sequences of pfs47 and p47rec available in GenBank were downloaded. Only sequences identified as belonging to Plasmodium spp and Anopheles spp were included in the search (Additional Material 1).

Pipeline used to generate functional and structural annotation predictions of the Pfs47-P47Rec complex. (A) Data mining and development of data sets; (B) data filtering; (C) selection of unique sequences; (D) evolutionary and phylogenetic analysis; (E) pattern recognition and primary protein structure prediction; (F) protein secondary structure prediction and mutations; (G) protein domain search; (H) identification of antigenic sites; (I) prediction of folding proteins; and (J) protein-protein interaction models.
Data mining and filtering
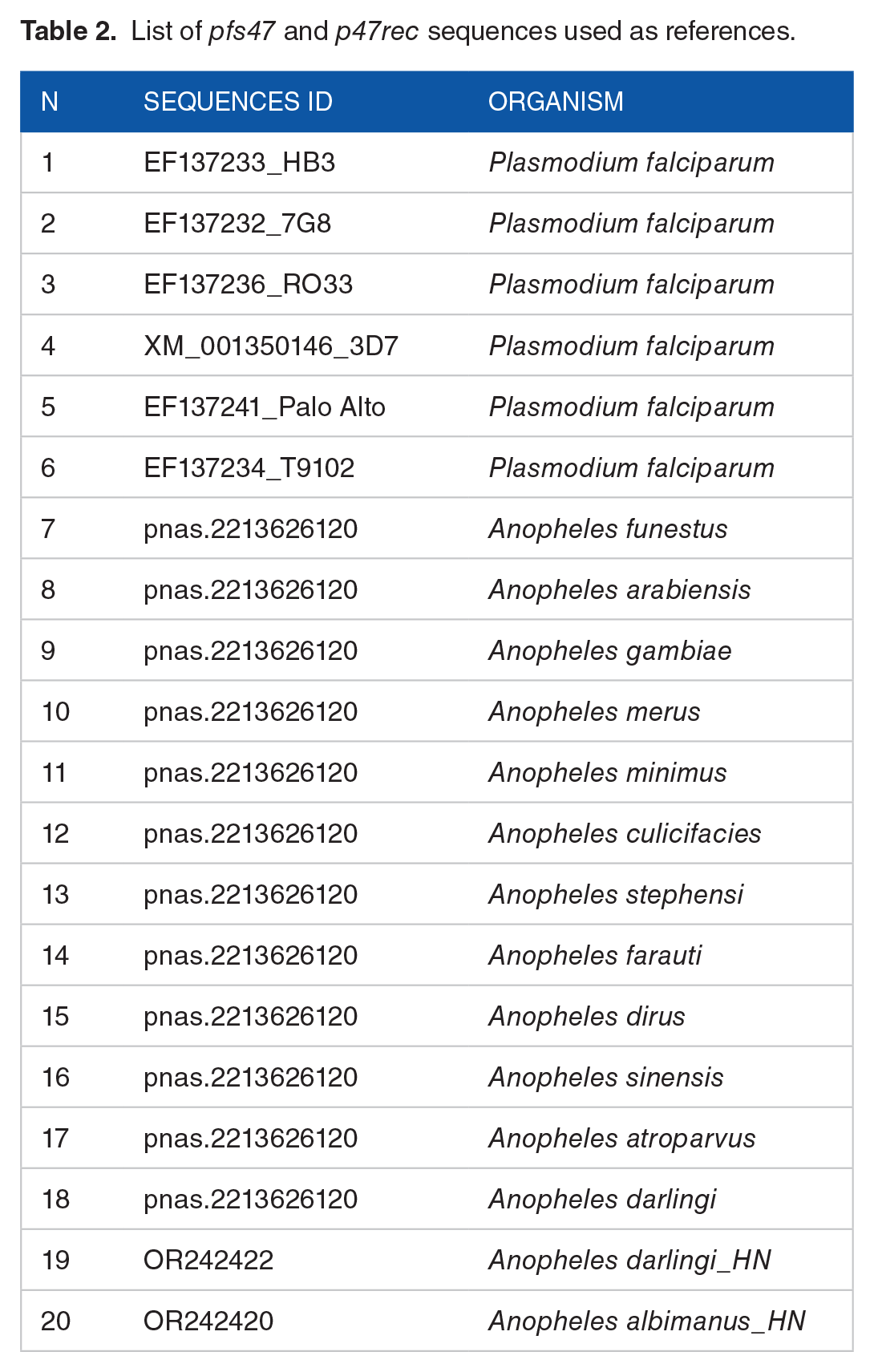
A data mining approach based on global multiple alignments of reference sequences was used. Reference sequences were used as ‘baits’ to identify and select sequences with a high level of similarity (Table 2). The sequences were selected and stored in 2 separate files, 1 for each of the 2 proteins of the complex. These sequences formed the initial data sets that were used in subsequent analyses.
List of pfs47 and p47rec sequences used as references.
Incomplete, fragmented, redundant or repeated sequences were filtered and deleted from the original data set. Filtered sequences were aligned with the reference sequences in AliView v.1.28 for Linux. 16 The resulting alignments were subjected to haplotype analysis using the R Haplotype package 17 to discard redundant sequences that had not been eliminated by the first filter, keeping unique sequences in the following steps. The resulting sequences were translated into amino acids using the ExPASy Translate Tool 18 and were subsequently aligned to identify and filter haplotypes with synonymous translations. Only reference sequences were retained for subsequent analyses, except for situations where reference sequences existed in haplotypes with multiple sequences. In other cases, a random sequence of each haplotype with multiple sequences was stored. Translated sequences were aligned again, which allowed filtering out the sequences that translated synonymous proteins.
Evolutionary and phylogenetic analysis
Phylogenetic trees were created using the amino acid sequences obtained in the previous analysis through the advanced mode of the Phylogeny.fr server. Trees were generated using sequences aligned with MUSCLE 19 in its full mode. Curation was performed with Gblock 20 (using the analysis model that prevents many contiguous positions from requiring consensus, using the server). For evolutionary inference and distance calculation, the maximum likelihood method with 100 bootstraps and the Jones-Taylor-Thorton (JTT) amino acid substitution model were selected. Finally, the results were visualized and edited in Figtree v.1.3.0 for Linux. Motif pattern identification analyses were performed using the MEME-Suite motif search function v.5.5.3 for Linux, 21 to evaluate the general architecture and infer the possible secondary structures of the proteins.
Pattern recognition and primary protein structure prediction
In addition to the search for motifs, an analysis was carried out to check whether the patterns could be related to the co-adaptation processes of both proteins of the complex, as well as their possible geographical relationship. JalView v. 2.11.2.6 for Linux was used to predict protein secondary structures using the default parameters for the JPred secondary structure predictor. 22 A comparison of the secondary protein structure of the Pfs47 and P47Rec sequences was performed by aligning the proteins in JPred. This analysis sought to determine the likelihood that the mutations could alter the secondary structure of the polypeptide at the places where they are present and thus impact the folding of the proteins.
Secondary and tertiary structure predictions and functional prediction of domains
SignalP v5.0 for Linux 23 was used to analyse the individual sequences of the Pfs47 and P47Rec proteins, to detect signal peptide-like domains. Simultaneously, the sequences were subjected to transmembrane domain identification analysis with TMHMM v2.0 software for Linux. 24 This was carried out to identify the presence of tertiary structures previously described for Pfs47.13,25 In addition, both data sets were subjected to analysis to reveal structures or domains not previously described. Subsequently, the sequences were subjected to tertiary structure prediction using the default settings of AlphaFold software v.2.3.2, 26 which can be accessed through the Colab Research Server of Google.
The tertiary structures were subjected to quality control in the web version of MolProbity software. 27 The predicted angles were evaluated using Ramachandran diagrams, to demonstrate the biological plausibility of the tertiary structures. The structures were modelled using BepiPred v3.0 software in its web version. 28 Antigenic sites with a greater probability of generating epitopes and, therefore, a greater probability of protein-protein interaction were identified.
Protein-protein interaction models
Tertiary prediction results were viewed and edited in UCSF-Chimera X v.1.6.1 for Linux. 29 Finally, 6 Pfs47 structures were subjected to docking analysis alongside 15 P47Rec structures. The Anopheles species subjected to P47Rec sequence analysis included A albimanus, A gambiae, A arabiensis, A coluzii, A merus, A funestus, A dirus, A farauti, A culicifacies, A marshalii, A moucheti, A aquasalis, A cruzii, A stephensi, A minimus, A sinensis, A bellator, A atroparvus and A darlingi from South America. Sequences obtained in this study from A darlingi and A albimanus collected in Honduras were included in the analysis. The Pfs47 models were generated based on the genetic sequences obtained from the following P falciparum strains: 3D7 (Africa), 7G8 (South America), HB3 (Honduras), Palo Alto (Africa), RO33 (Africa) and T9/102 (Asia). Sequences of plasmodia isolated from nonhuman primates were included. This analysis was performed to determine the level of affinity that these proteins could have with P47Rec and their phylogenetic relationship with other Pfs47 alleles of Plasmodium falciparum. This analysis was performed using the default HDOCK SERVER parameters, defining Pfs47 as the receptor and P47Rec as the ligand in the interaction model. 30 For this analysis, only the reference sequences of both proteins were used, mainly due to the massive amount of information generated.
To evaluate the results of the docking simulations, a comparison model was defined based on the following parameters: The final comparisons had to involve at least 2 models as an interaction reference. One of the models was in all cases the consensus protein P47Rec interacting with each Pfs47 receptor. On the contrary, the second reference model used was selected based on experimental evidence of the interactions between the proteins of the complex. This model was called the ‘ideal interaction model’ (IIM). For interaction models of parasite strains originating from Africa (RO33, 3D7 and Palo Alto), the IIM was ‘Pfs47 – A gambiae’. Regarding the American strain 7G8, the Pfs47-A darlingi model was used as an interaction model. For the HB3 model, the Pfs47-A albimanus model was used, while for Pfs47-T9/102, Pfs47-A sinensis was chosen. Finally, the reference interaction sites were delimited based on the positions of the IIM and the best model of the P47Rec consensus. For simulations producing multiple models with similar interaction sites, the selection of IIMs was based on the magnitude of the root mean square deviation (RMSD) of the atomic positions, taking as a reference IIM the model with the lowest RMSD.
Results
Sequencing of the p47rec gene of Anopheles albimanus and Anopheles darlingi from Honduras
Four sequences (two from each species) of the p47rec gene were obtained from A albimanus and A darlingi collected in Honduras. The sequences were deposited in GenBank with accession numbers OR242420 to OR242423. The analysis of the 2 A albimanus consensus sequences resulted in the identification of 7 single-nucleotide polymorphisms (SNPs), with a pairwise percentage identity of 99.5%. The 2 sequences of A darlingi exhibited 4 SNPs and a pairwise percentage identity of 99.7%. The comparison of p47rec nucleotide sequences between A albimanus and A darlingi revealed 92% identical sites and a pairwise percentage identity of 94.8%. When translating the sequences, 97.6% of the sites were identical, and a pairwise percentage identity of 98.4% was recorded.
Data mining and data set development
The data mining strategy generated a total of 128 and 134 nucleotide sequences for pfs47 and p47rec, respectively. Short or incomplete sequences, as well as redundant sequences, were aligned and discarded, obtaining a total of 57 sequences for pfs47 and 55 sequences for p47rec. Subsequently, the haplotypes for each protein were analysed, obtaining a total of 47 haplotypes for Anopheles spp and 57 haplotypes for P falciparum. As in the previous instance, random or reference sequences were conserved as appropriate for each case. Finally, a total of 24 unique sequences for Pfs47 and 40 sequences for P47Rec were obtained (Additional Material 2). These sequences were used to develop the final data sets that were subjected to subsequent analyses.
Evolutionary and phylogenetic analysis
The phylogenetic tree obtained for Pfs47 showed 3 distinct clusters. A first cluster named African sensu stricto (ss), a second cluster called Asian with sequences from parasites collected in Asia, along with Plasmodium species from other primates (named AOP), and a third cluster called ‘AAA’ which included African, Asian and American Pfs47 sequences. In all cases, Pfs47 clusters always included at least 1 strain of African origin. Pfs47 did not show clear clusterization based exclusively on the geographical origin of the parasites (Figure 2). On the contrary, the consensus sequence of Pfs47 showed similar evolutionary relationships with the African ss cluster and the AOP cluster. The AAA cluster, on the contrary, showed a closer relationship with the African cluster. No geographical association was observed in the AAA cluster.

(A) Phylogenetic tree for Pfs47 showing 3 clusters. Cluster 1: African sensu stricto (red), cluster 2 AOP: Asian with sequences of parasites collected in Asia, along with Plasmodium species from other primates (blue), and a third cluster called AAA (green). Light blue includes parasite sequences from nonhuman primates. (B) Phylogenetic tree for P47Rec showing 3 clusters. Cluster 1: African sensu stricto (red), cluster 2: Asian (blue) and a cluster including anophelines from America (green). The light blue includes sequences of South American anophelines. Yellow represents the consensus sequence in both cases (A and B).
On the contrary, the phylogenetic tree of P47Rec showed a distribution of clusters that corresponded to the geographical distribution of the anopheline species studied (Figure 2). The analysis identified 5 distinct clusters based on P47Rec: (1) an African cluster, where the consensus sequence of P47Rec was found, (2) an African-Asian cluster and (3) a Eurasian cluster, and 2 clusters (4 and 5) with species from the Americas. Sequences of 3 species of African origin were identified within the African-Asian cluster (A funestus, A marshalii and A moucheti), in addition to 5 species native to Asia (A dirus, A farauti, A culicifacies, A stephensi and A minimus), whereas the Eurasian cluster was formed by the sequences of A sinensis and A atroparvus. The fourth cluster comprised sequences of South American anophelines, specifically A cruzii and A bellator. In the fifth cluster, sequences of American species were organized into 3 subclusters. The first subcluster consisted of sequences of A albimanus from Honduras. The second subcluster included sequences of A darlingi from both Central and South America, as well as a sequence from A aquasalis. The third subcluster exclusively contained sequences of A aquasalis.
Identification of motif patterns and prediction of primary structures
All Pfs47 sequences displayed the same pattern, which was composed of 11 conserved motifs, except for the EF137234_T9/102 reference sequence, which displayed a distinct pattern at the N-terminus. The missing region in EF137234_T9/102 was later found to be located within the signal peptide domain (Figure 3). Although 11 motifs made up the basic pattern of Pfs47, the analysis identified a total of 14 different motifs, each with at least 1 potential variant (Additional Material 3).

(A) General pattern of amino acid motifs of the Pfs47 protein from P falciparum EF137234_T9/102. (B) Motifs of the P47Rec protein from Anopheles spp.
P47Rec, on the contrary, exhibited 10 different motif patterns and 15 conserved motifs. Only 1 motif was different between the number of identified motifs and conserved motifs. Pattern 1 was the most frequent (24/36 sequences) in P47Rec sequences from all continents (Figure 3) (Additional Material 4). Pattern 3 was the second most frequent pattern (3/26), and the 3 sequences included within this pattern presented geographical association. All sequences with pattern 3 belong to American anophelines and included sequences from A albimanus and A darlingi collected in Honduras, as well as A aquasalis.
Prediction of secondary structures and identification of mutations
The predictions of the secondary conformation of Pfs47 revealed a basic structure made up of 11 alpha helices and 22 beta sheets, arranged as depicted in Figure 4. The N-terminal region of Pfs47 revealed a signal peptide-like domain between amino acids 1 and 18, as well as a cleavage site between amino acids 18 and 19. The secondary structure of the proteins also revealed a putative GPI-type anchoring site and a transmembrane domain close to the C-terminus. Ninety-one mutation sites or 21% of the amino acids that make up all proteins were found. The transmembrane domains and the signal peptide were unaffected, and the mutations were limited to 6 locations in the protein’s central globular domain. Substitutions involving valine (V), leucine (L), phenylalanine (F), methionine (M) or isoleucine (I) occurred in 24 of the 91 mutations (26%) (Additional Figure 1). Furthermore, glutamic acid (E) was replaced by glutamine (Q), aspartic acid (D) or lysine (K) in 15 of the 91 mutations (16%). The remaining Pfs47 mutations were not associated with commonly observed substitution patterns.

Secondary structure predictions of (A) Pfs47 and (B) P47Rec. Red cylinders visually represent Alpha helices, beta sheets are denoted by green rectangles, and loops are depicted as grey lines. Asterisks show the location of the amino acid substitutions that had the greatest effects on secondary structure predictions; while the dotted boxes show the regions that underwent significant structural changes.
The secondary structure of P47Rec was predicted, revealing a minimum of 20 beta sheets (Figure 4). Moreover, it was predicted that P47Rec lacks transmembrane domains, signal peptides and GPI anchor sites. Even so, the alignments showed proteins exhibiting conserved secondary structures and mutations that predominantly impact amino acids possessing chemically similar characteristics (V, L, F, M and I). A total of 47 mutations were identified in the secondary structures of P47Rec, which represent approximately 16% of the amino acids. The remaining mutations did not exhibit discernible substitution patterns, except for those involving hydrophobic amino acids (E, Q, D and K) (Additional Figure 2).
The Pfs47 consensus sequence was subjected to secondary structure prediction, which revealed 3 domains previously described. 25 The composition of this structure consisted of 3 alpha helices and 24 beta sheets (Figure 4). These findings show agreement between the Pfs47 consensus sequence, and the secondary structure prediction calculated by Jpred, whereas the secondary structure prediction of the P47Rec consensus sequence revealed an even distribution of 28 beta sheets throughout 2 protein domains (Figure 4). Moreover, the analysis of P47Rec did not reveal any evidence of signal peptides, transmembrane domains or signalling and membrane anchoring structures.
Prediction of tertiary structures and identification of antigenic sites
To predict the tertiary structure of Pfs47, the removal of the signal peptide from the sequences was performed by the cleavage site. This process was conducted by manually removing the signal peptide sequences from the unique sequences subjected to structural prediction. Tertiary structures unveiled a protein with 2 distinct domains. The first domain was a transmembrane domain, characterized by a helical structure consisting of 20 amino acids. The second domain was globular, taking the form of an equilateral triangle. This domain comprises 3 substructures arranged in a sandwich-like manner, with each substructure composed of an average of 8 antiparallel beta sheets (Figure 5). The globular domain of Pfs47 exhibits sandwich-like structures at both its C- and N-terminal ends. These structures belong to the 6-Cys family and contain 3 disulfide bonds in the central region (Additional Figure 3). On the contrary, the central substructure did not exhibit any discernible functional annotation (Additional Figure 4). Nonetheless, it is notable that the central substructure has 2 cysteines in proximity. However, it appears that these cysteines do not correlate directly with the substructure’s rigidity (Additional Figure 4). According to our substitution models, the region containing the cysteine residues in the core substructure did not exhibit notable changes when substituted with glycine. This suggests that the cysteines may not contribute to the rigidity of the subunit, implying that this region might have a role in the specificity of the interaction with P47rec.

Tertiary structure predictions of (A) Pfs47 and (B) P47Rec. Row 1 of each model presents the cartoon structure. Row 2 shows the volumetric models. Row 3 presents the electrostatic forces. Row 4 shows the hydrophobicity. Columns B, C and D present the models with rotations of 90°C in the x-axis concerning column A; columns E and F present the models with rotations of 180°C in the y-axis concerning column A.
The tertiary structure of the Pfs47 consensus sequence exhibited regions on the upper surface of the globular domain that are less electrostatic towards the apex and more electrostatic towards the base (relative to the triangular structure of the domain) (Figure 5). In contrast, the lower face of the globular domain exhibited fewer electrostatic areas at the ends, while displaying highly electrostatic regions in the central region of the domain. Electrostatic amino acids predominated in 3 of the 4 lateral faces of the protein. Moreover, the transmembrane domain of Pfs47 predominantly comprises lipophilic regions, which is consistent with the predicted characteristics of this domain. In contrast, the globular domain exhibits predominantly hydrophilic regions throughout its surface (Figure 5).
The findings derived from the analysis of the Ramachandran plots showed few improbable angles, and in most cases, these represented less than 1% of the total angles between amino acids. These angles were generally located near the inclusion zones in the plots in the consensus sequences (Figure 6). The consensus protein P47Rec revealed a tertiary structure that simulates a dumbbell (Figure 6) and has 2 globular domains of the DM9 repeat type (Additional Figure 3), separated by an interdomain loop (Figure 5). Only a small portion of P47Rec showed electrostatic potential, while the rest of the protein surface exhibited regions with low electrostatic potential. A considerable proportion of the protein exhibits regions with hydrophilic properties; however, the hydrophobicity pattern does not reveal notably hydrophobic segments (Figure 5).

(A and C) Location of cysteine residues in the structures of the Pfs47 and P47Rec consensus proteins. (B and D) Ramachandran plots of the consensus proteins of Pfs47 and P47Rec.
Pfs47 showed 18 antigenic sites, among which the sites designated II, III, VI, X, XI, XI and XVI had the greatest abundance of amino acids (Additional Figure 5). The antigenic sites were later identified and defined inside the tertiary structures that were constructed using Alphafold. The visualization of these data was performed using UCSF-ChimeraX. When comparing the Pfs47 consensus structure to the triangle structure, it was observed that the antigenic areas were predominantly situated at the lower and upper right vertices (Additional Figure 6). In contrast, the P47Rec sequences exhibited a total of 10 antigenic regions, which were evenly distributed across the N- and C-terminal domains. Notably, regions I, IV and VII encompassed the highest number of amino acids (Additional Figure 5). As in the case of Pfs47, not all antigenic sites were located on the surface of the folded protein. For the P47Rec consensus, a region close to the interdomain loop with 3 antigenic sites was observed (Additional Figure 6). The N-terminal domain of the P47Rec consensus protein exhibited a greater number of antigenic sites on its front face compared with the C-terminal domain, which had only a small number of antigenic sites.
Pfs47-P47Rec interaction models
A total of 8400 PDB files containing the top 100 best molecular docking models of the Pfs47-P47Rec complex were analysed. All generated models were filtered, selecting only those models that presented RMSD less than 40 Å. Then, the best models of each docking were displayed, and only those interaction models that involved the upper face of Pfs47 were filtered. A total of 686 interactions were chosen for further analysis. In the absence of an existing method to assess this particular analytical approach, we aimed to develop evaluation criteria that would enable an objective assessment of the generated models. The evaluation focused on specific metrics, including interaction distance, similarity with the consensus model and IIM, and the geographical relationships between the Pfs47-P47rec complexes and their relationship with the specificity of each possible model. The overall average RMSD of the selected models was 35.92 Å (Additional Material 5). The models exhibiting the lowest RMSD for each docking process were chosen. These selected models were then visually examined and compared with the remaining models for each Pfs47 receptor (Figure 7). For the Pfs47-RO33 interaction, the reference models showed diametrically different positions, while the consensus model showed P47Rec located in the centre of the upper face of Pfs47 and tilted to the left. P47Rec from A gambiae showed an interaction zone more related to the N-terminal substructure of Pfs47-RO33 and was also inclined in the opposite direction. Apart from the following 3 models: Pfs47-RO33-A stephensi, Pfs47-RO33-A funestus and Pfs47-RO33-A merus, all interaction models resembled the location of the P47Rec consensus sequence. Pfs47-RO33-A merus was the model with the greatest similarity to the IIM of Pfs47-RO33 (Figure 8) (Additional Material 6).
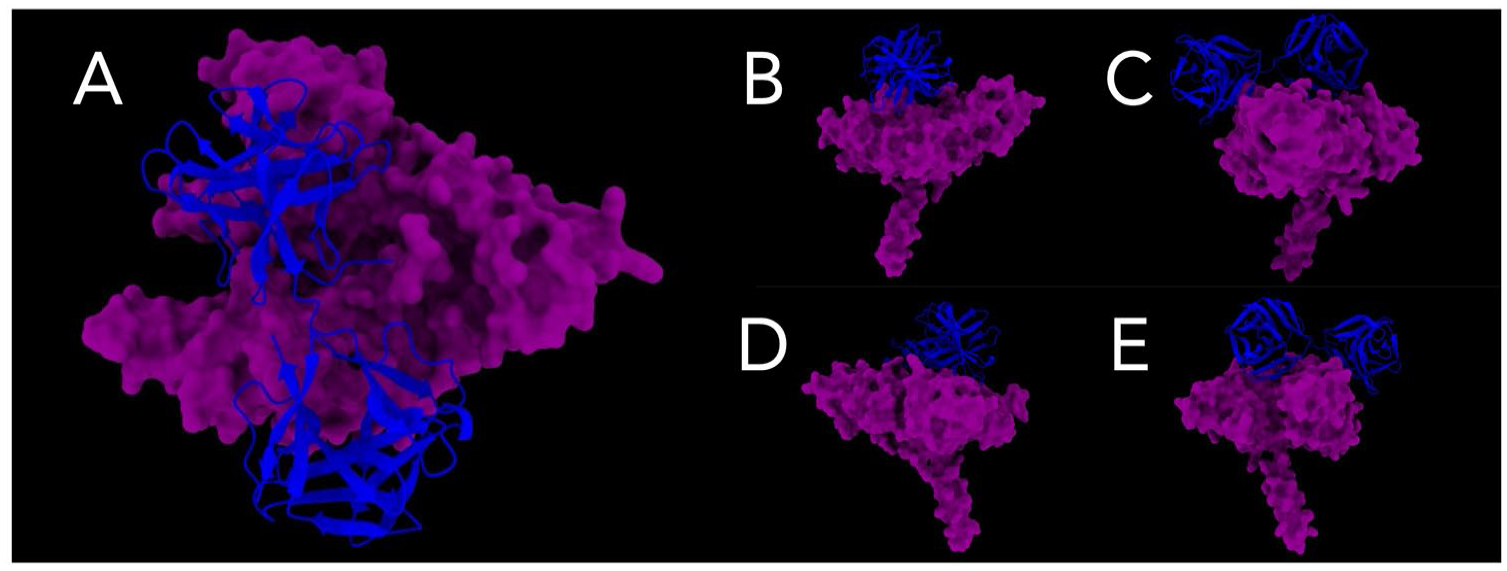
Interaction models between Pfs47-P47Rec. (A) The upper face of the interaction site between the consensuses is depicted. (B) to (E) provide lateral views of the consensus interaction site.

Optimal docking models for the complexes formed by the interaction of Pfs47 and P47Rec. (A) Pfs47-RO33, (B) Pfs47-3D7, (C) Pfs47-Palo Alto, (D) Pfs47-7G8, (E) Pfs47-HB3 and (F) Pfs47-T9/102.
Regarding Pfs47-3D7, it was observed that both the consensus and the IIM were situated in similar locations and had identical orientations. With the exclusion of models Pfs47-3D7-A culicifacies, Pfs47-3D7-A arabiensis and Pfs47-3D7-A funestus, the remaining models exhibited similarities in the regions of interaction (Figure 8). In contrast, in the Pfs47-Palo Alto model, both the P47Rec consensus and the IIM were observed to be situated in comparable locations, but with slight variations in their inclinations. Except for Pfs47-Palo Alto-A darlingi (from South America) and Pfs47-Palo Alto-A culicifacies, all models resembled the P47Rec consensus or the IIM. The models Pfs47-Palo Alto-A arabiensis and Pfs47-Palo Alto-A merus had the highest degree of similarity in their location and orientation in relation to the IIM for this receptor (Figure 8). In contrast, the reference models of Pfs47-7G8 showed different locations and orientations. One interesting characteristic of the P47Rec consensus sequence is its leftward inclination, which is situated within the central region of Pfs47-7G8. In contrast, the IIM exhibits a virtually vertical positioning between the left vertices of Pfs47-7G8. For this docking, the following 7 models were similar to the IIM: Pfs47-7G8-A albimanus, Pfs47-7G8-A darlingi (from Honduras), Pfs47-7G8-A atroparvus, Pfs47-7G8-A minimus, Pfs47-7G8-A arabiensis, Pfs47-7G8-A funestus and Pfs47-7G8-A merus. The remaining models did not exhibit any noticeable patterns (Figure 8).
In the Pfs47-HB3 model, it was observed that the P47Rec consensus and the IIM were not consistent in position or orientation. Using the Pfs47-HB3-A albimanus model as a reference, only Pfs47-HB3-A minimus and Pfs47-HB3-A culificacies showed similarity in location and orientation in relation to the IIM, while the following 5 models presented a location and orientation more similar to the P47Rec consensus model: Pfs47-HB3-A darlingi (from South America), Pfs47-HB3-A sinensis, Pfs47-HB3-A dirus, Pfs47-HB3-A gambiae and Pfs47-HB3-A arabiensis. The other models did not show significant similarities with any of the comparison models (Figure 8). Finally, neither the P47Rec consensus model nor the IIM for Pfs47-T9/102 coincided in location or orientation. When the model of Pfs47-T9/102-A sinensis was used as IIM, similarities were evident only with the models Pfs47-T9/102-A gambiae, Pfs47-T9/102-A dirus and Pfs47-T9/102-A merus, while the consensus P47Rec showed similarities in location and orientation with Pfs47-T9/102-A albimanus, Pfs47-T9/102-A atroparvus, Pfs47-T9/102-A minimus and Pfs47-T9/102-A stephensi. None of the remaining models had any similarities with the reference models (Figure 8).
Discussion
The findings of our study indicate that the Pfs47 derived from African strains of the parasite, as well as those from nonhuman primate parasites, have the highest similarity to the Pfs47 consensus sequence. Although the consensus is not a prediction of ancestry, this sequence was located equidistant from all the clusters, so its structure is representative of all other proteins. However, it is advisable to expand the databases to determine more precise evolutionary structures. This outcome could be attributed to the African origin of P falciparum and the spread from nonhuman primates to humans.31,32 However, to substantiate this hypothesis, it will be essential to increase the number of sequences analysed. This is supported by its placement within the cluster referred to as AOP (Figure 2) and its equidistant position relative to the African cluster ss and the consensus sequence.
On the contrary, the AAA cluster exhibited a greater relationship with the African cluster ss, thus adding support to the hypothesis that the first dispersal of P falciparum occurred from Africa to Asia. 9 In addition, the sequences that fall inside the AAA cluster exhibit evolutionary relatedness, implying a recent emergence in comparison with other Pfs47 variants grouped in other clusters. This finding provides more support for the existing information about the transmission of P falciparum from Africa to the Americas during the period of European colonization. 33 Considering the genetic distances between the Pfs47 variants and the consensus sequence, it is possible to argue that some proteins such as Pfs47-Palo_Alto and Pfs47_T9/102, due to their proximity to the consensus protein and the cluster of other primates, could have emerged sooner. Although the consensus sequence is not an ancestral protein, its location in the tree makes it intermediate between the clusters. To test this hypothesis, it will be necessary to include new sequences of these proteins to calculate ancestry with greater precision. While this study found that a significant portion of the nucleotide sequences can be translated into synonymous proteins, it is possible that increasing the number of sequences would not have a substantial impact on the overall structure of the phylogenetic trees of the protein complexes. Conversely, Pfs47-7G8 and Pfs47-HB3 from Brazil and Honduras, respectively, may represent more recently evolved receptor proteins.
The anophelines of the Old and New World exhibit significant evolutionary divergence, occurring approximately 95 million years ago. This divergence coincides with the separation of the South American continent from its present-day African continent. 34 These results could indicate a sequential dispersion of P47Rec from Africa to other geographic regions. However, to verify this hypothesis, it is still necessary to expand the proteins analysed, including those from other anopheline species. 35 The phylogenetic tree based on the P47Rec sequences shows sequential clusters that go from Africa to Asia, Eurasia, South America and Central America, suggesting that the speciation of South American and Central American anophelines is more recent.8,36 In contrast, the intermediate cluster positioned between the Asian cluster and the American subcluster comprised the reference sequence of A atroparvus from Europe, thereby shedding light on the interconnections between Eurasian anophelines. 37
The structure of the phylogenetic tree of P47Rec demonstrates the presence of diversity across the anopheline species that were analysed. This observation may be associated with the geographical spread of the genus, as well as with vector speciation. According to the motif patterns of Pfs47, a highly conserved protein structure is observed, as described by other authors.13,25 Considering the role of Pfs47 in evading the immune system of its vectors, it is reasonable to expect that a significant part of its structure exhibits a notable level of conservation.
The Pfs47-T9/102 sequence exhibited a distinct motif pattern in the signal peptide region, which differed somewhat from the patterns observed in other evaluated polypeptides. Based on the available evidence, it can be argued that the observed variation in the Pfs47-T9/102 pattern is unlikely to have any significant effect on the protein’s interaction capacity. Our analysis of the annotations indicated that both the motif pattern of Pfs47-T9/102 and that of the rest of the proteins possess a cleavage site that would remove the signal peptide from all proteins.
On the contrary, P47Rec has a higher abundance of motif patterns compared with Pfs47. Only pattern 3 encompasses species exclusively from a single continent. The remaining patterns encompassed an assortment of anopheline species originating from various geographical locations.
The primary variations in the configurations of P47Rec occur mainly in the C-terminal region, indicating that this segment may play a crucial role in the specific interaction with Pfs47. This finding lends support to the hypothesis proposing coevolutionary associations between Pfs47 and P47Rec, dependent on their geographical locations.9,25,37
The mutations found in the primary structures of Pfs47 and P47Rec have the potential to impact protein folding, particularly when these mutations involve amino acids with different charges. The structure of proteins can be modified by changes in charges and the lengths of the side chains of amino acids, which can lead them to reject one other due to their polarities or the volume they occupy. A greater number of mutations would generate a greater number of structural modifications. Nevertheless, the secondary and tertiary structures of the examined sequences, along with the consensus sequence, exhibited similar basic structures. The observed similarities may indicate that slight structural differences in the complex account for the selectivity observed between P falciparum strains and their respective vectors in different geographical areas.9,37
Concerning tertiary structures, it was shown that Pfs47 exhibits 3 sandwich-shaped structures within its globular domain, which are likely responsible for its interaction with P47Rec. 13 A few cysteine residues in the globular domain of Pfs47 at its C and N-terminal ends imply a potential association with the protein’s rigidity. Conversely, the central structure of Pfs47 contains only 2 cysteines, indicating a possible involvement of this region in the induced adjustments during the formation of the complex 13,37,38 confirming the prediction of a loop structure at this site, likely involved in binding to P47Rec. 8
The lack of membrane anchoring structures is noteworthy, as observed in P47Rec. This result suggests that P47Rec could be a subunit of a larger protein complex that provides the necessary structures to anchor to the membrane, but a different hypothesis supports the findings of Molina-Cruz et al, proposing that P47Rec may exhibit cytoplasmic localization and have a function in the arrangement of the cytoskeleton within the midgut cells of mosquitoes. 8
The prediction of interactions between the consensus sequences of both proteins shows more precise interactions between the 6-Cys structures of Pfs47 and the N-terminal domain of P47Rec. Interactions between the central substructure of the globular domain of Pfs47 and some models of P47Rec were observed, suggesting a potential role for the N- and C-terminal regions of Pfs47 and the N-terminal end of P47Rec in mediating the primary interaction within the complex. Nevertheless, it seems likely that the central structure of Pfs47 and the C-terminal end of P47Rec exhibit distinct interaction regions, potentially accounting for their respective specificities.9,13,37 Some interaction models of the complex, such as those observed in the Pfs47 receptors of American strains of the parasite and the P47Rec variants of American anophelines, support the hypothesis of more recent coevolution. 25 It is noteworthy that a majority of the assessed docks of the Pfs47-P47rec complex with geographical proximity exhibited similarities to their respective IIMs or consensus models. This finding implies that the coevolution of Pfs47 and P47rec may have led to particular interactions between Anopheles species and regional strains of P falciparum.
Overall, the study’s novelty lies in its integrative approach combining bioinformatics, phylogenetics, structural biology and region-specific data collection to provide a comprehensive understanding of the coevolution and interaction between Pfs47 and P47Rec. This research not only fills gaps in current knowledge but also paves the way for future studies to explore these complex biological interactions further.
Conclusions
The findings of our study indicate that there are coevolutionary patterns between P47Rec and Pfs47 associated with the speciation and geographic dispersion of Anopheles spp and P falciparum. Furthermore, the evidence indicates that the evolutionary variations of Pfs47 have occurred more recently than those of P47Rec. This suggests a potential correlation between the parasite’s adaptability to existing anopheline species in different regions of the world. Our findings indicate that P47Rec lacks the necessary structural elements for membrane attachment, which suggests a cytoplasmic localization or a more complex structural organization. A potential molecular model between Pfs47 and P47Rec is revealed by the delineation of the complex’s potential interaction regions and the dockings generated through the model sequences. Although our results demonstrate statistically probable simulations, they do not necessarily reflect the actual interaction of the complex system. The validity of these findings might be further confirmed through the utilization of structural biology methodologies, like cryo-electron microscopy (cryo-EM). 39 A major limitation of our study is the scarcity of sequences for both P47Rec and Pfs47 in global databases. This restricts the ability to develop more precise interaction models. It is advisable to continue the study of these molecular markers in parasite isolates from various geographical areas, as well as other anopheline species, to increase the number of sequences and thus improve the understanding of the structure and interaction between these proteins. This study not only enhances our understanding of the Pfs47-P47rec complex but also introduces a method to assess molecular docking models using coevolutionary techniques. This research not only enhances our understanding of how the immune system of malaria vectors evades detection but also demonstrates the use of structure prediction as a tool to analyse coevolutionary systems at the molecular level.
Supplemental Material
sj-png-1-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-1-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Supplemental Material
sj-png-2-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-2-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Supplemental Material
sj-png-3-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-3-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Supplemental Material
sj-png-4-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-4-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Supplemental Material
sj-png-5-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-5-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Supplemental Material
sj-png-6-bbi-10.1177_11779322241284223 – Supplemental material for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach
Supplemental material, sj-png-6-bbi-10.1177_11779322241284223 for Coevolutionary Analysis of the Pfs47-P47Rec Complex: A Bioinformatics Approach by Andrés S Ortiz-Morazán, Marcela María Moncada, Denis Escobar, Leonardo A Cabrera-Moreno and Gustavo Fontecha in Bioinformatics and Biology Insights
Footnotes
Acknowledgements
Not applicable.
Author Contributions
GF and ASO-M involved in conceptualization. DE, GF, ASO-M, LAC-M and MMM involved in methodology. ASO-M, GF and DE involved in validation. ASO-M and GF involved in formal analysis. DE, GF, ASO-M and LAC-M involved in investigation. GF involved in resources. ASO-M, GF and DE involved in data curation. ASO-M and GF involved in writing – original draft preparation. DE, GF, ASO-M, LAC-M and MMM involved in writing – review and editing. GF involved in supervision. DE involved in project administration. GF involved in funding acquisition.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: The National Autonomous University of Honduras’ Genetic Research Center provided funding for this project.
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Materials
The data sets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.