
Abstract
CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) is a naturally occurring genetic defense system in bacteria and archaea. It is comprised of a series of DNA sequence repeats with spacers derived from previous exposures to plasmid or phage. Further understanding and applications of CRISPR system have revolutionized our capacity for gene or genome editing of prokaryotes and eukaryotes. The CRISPR systems are classified into 3 distinct types: type I, type II, and type III, each of which possesses an associated signature protein, Cas3, Cas9, and Cas10, respectively. As the CRISPR loci originated from earlier independent exposures of foreign genetic elements, it is likely that their associated signature proteins may have evolved rapidly. Also, their functional domain structures might have experienced different selective pressures, and therefore, they have differentially diverged in their amino acid sequences. We employed genomic, phylogenetic, and structure-function constraint analyses to reveal the evolutionary distribution, phylogenetic relationship, and structure-function constraints of Cas3, Cas9, and Cas10 proteins. Results reveal that all 3 Cas-associated proteins are highly represented in the phyla Bacteroidetes, Firmicutes, and Proteobacteria, including both pathogenic and non-pathogenic species. Genomic analysis of homologous proteins demonstrates that the proteins share 30% to 50% amino acid identity; therefore, they are low to moderately conserved and evolved rapidly. Phylogenetic analysis shows that 3 proteins originated monophyletically; however, the evolution rates were different among different branches of the clades. Furthermore, structure-function constraint analysis reveals that both Cas3 and Cas9 proteins experiences low to moderate levels of negative selection, and several protein domains of Cas3 and Cas9 proteins are highly conserved. To the contrary, most protein domains of Cas10 proteins experience neutral or positive selection, which supports rapid genetic divergence and less structure-function constraints.
Introduction
The ability of clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 to easily program the system provides an exciting gene-editing technology capable of editing the genomes of both prokaryotes and eukaryotes, including the human genome. The CRISPR/Cas9 system has been successfully applied to many different types of cells and organisms, such as bacteria, fungi, viruses, parasites, plants, animals, and human cell lines. 1 In addition, the system has been successfully employed to create transgenic animals. 2
The CRISPR is a naturally occurring genetic defense system in bacteria and archaea. 3 The CRISPR locus has a unique DNA sequence structure consisting of direct repeats, ranging from 21 to 37 nucleotides, interspaced by non-repetitive sequences of similar size. 4 The CRISPR defense has 3 separate phases: adaptation, expression, and interference. During adaptation, a short DNA fragment is removed from an invasive DNA and is incorporated into the CRISPR array in a site-specific manner to create a new spacer. In the second step, transcription of the CRISPR array results in a precursor CRISPR RNA (pre-crRNA) that binds with Cas proteins to undergo additional processing into mature crRNAs. Finally, during interference, the combined activity of crRNAs and Cas proteins recognizes and seeks the newly invasive DNAs and destroys the target nucleic acids. 5 The CRISPR systems are extensively distributed across the genomes of 42% of bacteria and 85% of archaea. 6 The CRISPR array matches the phage sequences that commonly invade bacteria. Previous studies corroborated that the non-repetitive spacers served as templates to target invading bacteriophages following previous exposures.7-9 The CRISPR systems were dependent on DNA complementary pairing because if the spacer in the CRISPR locus was no longer complementary to the phage genome, the systems could not seek and destroy the newly infecting bacteriophages. 7 Furthermore, the CRISPR systems were discovered to be transferable from bacteria with naturally occurring CRISPR systems to those lacking a CRISPR system by horizontal gene transfer. 4 The CRISPR systems also require cas genes, which encode CRISPR-associated proteins for the functionality of the system. 8
The CRISPR-Cas systems are classified into 2 major classes, class 1 and class 2, based on the number and protein composition participating in nucleic acid interference. These classes are further divided into 6 types and 33 sub-types, with multi-Cas protein effector complexes in Class 1 systems (types I, III, and IV) and a single effector protein in class 2 systems (types II, V, and VI).9,10 The signature proteins for types I, II, and III are Cas3, Cas9, and Cas10, respectively, and they perform different functions within their respective CRISPR systems as summarized in Table 1.
Comparison of Cas proteins: Cas3, Cas9, and Cas10.

Cas3 is an ATP-dependent single-strand DNA (ssDNA) translocase/helicase that is linked to an HD-nuclease domain (histidine-aspartate [HD] nuclease domain) in many CRISPR systems. During the part of a process known as “CRISPR interference,” the Cas3 translocase and nuclease activities break down DNA by reducing it to shorter fragments of tens of nucleotides, abolishing invading DNA.12,22,23
Cas9 is present in Eubacteria that cleaves viral DNA by unwinding and complementary pairing to the guide RNA and defend them against bacteriophages and plasmids.5,24 Apart from that, Cas9 can recruit proteins to a target site enabling a powerful engineered sequence-specific gene-editing and gene regulation control mechanisms.25,26 The multifunctional role of Cas9 is comprised of a more complex set of domains; however, the most important distinction is in its 2 nuclease domains, HNH and RuvC, which selectively cleave the target DNA. The dual role of Cas9 as a nuclease and an interferase lends to its simplicity for diverse genetic applications. The best characterized CRISPR system, type II CRISPR/Cas9, is a member of the class II system because it requires only the protein Cas9 for endonuclease activity. 27
Cas10 proteins belonging to type III contain an N-terminal HD-nuclease domain, 2 PALM domains separated by a zinc-finger motif (ZF), and a C-terminal domain (CT).28,29 It is thought that single-stranded DNase activity, observed for the HD domains of certain Cas10 proteins, promotes the nicking of ssDNA created during transcription.30,31,32 Many Cas10 proteins lack an N-terminal HD domain, and the quantity and phylogenetic distribution of such truncated proteins have not been thoroughly evaluated.6,33
The high sequence similarity within type III CRISPR systems as well as their similarity with type I systems suggest that type I may have evolved from type III. 34 In addition, type I and III systems use homologous HD-nuclease domains for catalysis.1,12,24 Furthermore, the multiprotein effector complexes of type I and III systems are more different from the single-protein activity of type II. The primary differences are due to the replacement of the HD-nuclease activity of the types I and III with the HNH and RuvC nuclease domains of the Cas9 protein of type II system. 22 The Cas9 protein of type II show no structural and functional similarity to any other proteins found in type I and III systems. 34 However, Cas9 appears to belong to a family of proteins that contains some protein family members in the type I system. It has been further indicated that the type II system originated from the fusion of the Cas9 transcript with the CRISPR locus from an unknown type I system. 8 The functional similarities between type I and II systems, such as the requirement of a PAM (protospacer adjacent motif) sequence, however, are inconsistent due to the lack of primary sequence similarity between Cas9 and other type I proteins.8,34 Therefore, even in the lack of primary sequence similarity, Cas9 and type I proteins may overall conserve their secondary and tertiary structure, which together contribute toward their similar function.
This study aimed to understand the protein homology, sequence conservation, evolutionary relationships, and structure-function constraints among Cas3, Cas9, and Cas10, employing amino acid similarity search using blastp, 35 phylogenetic method using a maximum-likelihood method, 36 and estimating the ratios of the non-synonymous substitution rate (dN) and the synonymous substitution rate (dS) per site between the 2 homologous protein sequences using MATLAB. 37
Materials and Methods
Bacterial genomes
The National Center for Biotechnology Information (NCBI) provides a collection of databases and bioinformatics tools for genome analysis. The nucleotide and protein databases were used from bacterial genomes which were completely sequenced and fully annotated. We examined 3 CRISPR-associated Cas protein families by identifying Cas protein homologs, phylogenetic analysis, and structure-function constraint analysis as summarized in the schematic diagram in Figure 1.
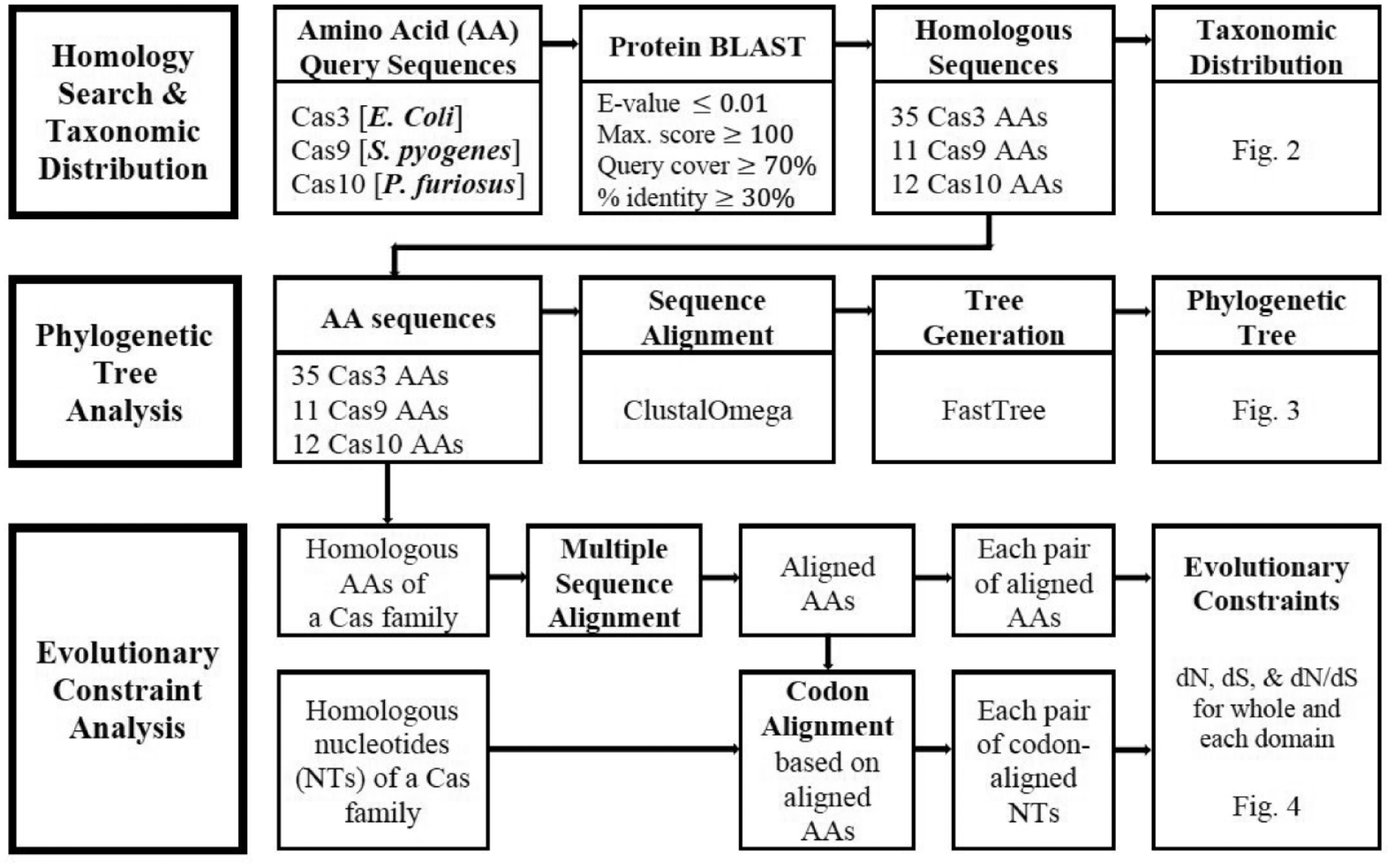
Workflow of homology search and taxonomic distribution, phylogenetic tree analysis, and evolutionary constraint analysis.
Identification of Cas protein homologs
The 3 Cas proteins, Cas3, Cas9, and Cas10, were examined in this study. Based on their common usage in previous studies of the CRISPR/Cas system, Cas3 of Escherichia coli, 38 Cas9 of Streptococcus pyogenes, 39 and Cas10 of Pyrococcus furiosus 40 were chosen as queries for DNA and non-redundant protein sequence similarity searches against the corresponding databases available at the NCBI using blastp. 35 The criteria for the homology search were as follows: E-value 10-2, Maximum Score 100, Query Coverage 70%, and percent amino acid identity >30%. From the filtered homologous protein sequences, only 1 amino acid sequence from each bacterial species was downloaded. Species were classified into phyla, such as Actinobacteria, Cyanobacteria, Firmicutes, Proteobacteria, or Spirochaetes.
To perform both the phylogenetic analysis and the structure-function analysis, the identified homologs were further reduced based on the availability of the corresponding nucleotide sequences in the NCBI database. The numbers of resulting selected sequences for Cas3, Cas9, and Cas10 were 35, 11, and 13, respectively. All the 59 sequences are available on request.
Phylogenetic analysis
Phylogenetic analysis was performed using the Geneious Prime platform, 41 which consists of several different tools and plugins for molecular sequence analyses. First, a total of 58 proteins multiple protein sequences were directly downloaded to the Geneious and then aligned using a multiple-sequence alignment method, Clustal Omega 42 with a default setting in the Geneious platform. Clustal Omega is based on seeded guide trees and Hidden Markov model (HMM) profile-profile techniques to generate multiple alignments. Then, an unrooted phylogenetic tree was constructed using an approximate maximum-likelihood method, FastTree 43 with a default setting in the Geneious platform. Unlike usual approaches that store distance matrices, FastTree stores sequence profiles of tree’s internal nodes and uses varied heuristics to quickly infer maximum-likelihood phylogenies for large number of sequences. The numbers above the branch points indicate the reliability of each split in the tree and these local support values estimated with the Shimodaira-Hasegawa test, in line with the SH-like local supports in PhyML3.0. 44 The tree is not drawn to scale; thus, branch lengths do not measure the number of substitutions per site.
Selective constraint analysis
For the constraint analysis for each of the 3 Cas families (Cas3, Cas9, and Cas10), a progressive multiple alignment (using multialgn function in MATLAB 37 ) was applied, and both non-synonymous substitution rate (dN) and non-synonymous substitution rate (dS) between 2 homologous nucleotide sequences (using dnds function in MATLAB) were estimated. The ratio (dN/dS) of these mutation rates along the pair of sequences was used to predict the selective constraints between the 2 entire protein sequences in each system as well as each different protein domain within each system. The separation of different domains allows the evolutionary constraints separated by the regions of the protein that are critical for functionality. For each Cas family, evolutionary constraints were analyzed for the entire length of sequences as well as the sequences corresponding to specific domains. As the amino acid residue numbers of Cas3 domain borders of Thermofibida fusca 13 were available, the domain information of T fusca was used to align and map to the corresponding domains of the Cas3 protein of E coli. The coordinates of the Cas9 domain borders of S pyogenes17,45 were used to identify the corresponding domain locations in 11 aligned Cas9 sequences. The coordinates of the Cas10 domain borders of Pyrococcus furiosus20,21 were used to identify the corresponding domain location in 12 aligned Cas10 sequences.
Note that the total of n(n − 1)/2 pairs can be made from n sequences in a Cas protein family. However, if the 2 sequences in a pair are too short, too divergent, or contain frame shifts, then saturation can be reached and the constraint values are unable to be estimated, resulting in Not-a-Number (NaN) values. Therefore, only valid constraint values estimated by dnds in MATLAB are used for the analysis. Each error bar summarizes evolutionary constraint values estimated for a specific domain in a Cas protein family, where the bar and the error represent the arithmetic mean and the standard deviation of the constraint values, respectively.
Results and Discussion
Distribution of Cas proteins across bacterial phyla
The percentage distribution of Cas3 protein in Proteobacteria, Firmicutes Actinobacteria, Bacteroidetes and other phyla are 29%, 20%, 9%, 9%, and 33%, respectively. The percentage distribution of Cas9 protein in Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and other phyla are 33%, 26%, 3%, 12%, and 26%, respectively. The percentage distribution of Cas10 protein in Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and other phyla are 23%, 20%, 3%, 11%, and 43%, respectively. Although the distribution of Cas3, Cas9, and Cas10 proteins among 3 phyla (Proteobacteria, Firmicutes, and Actinobacteria) are similar, significant differences exists among in the category of phylum Bacteroidetes and other phyla. The asymmetric distribution of Cas proteins in bacterial phyla is validated by a recent study where uneven distribution of CRISPR-Cas types was also reported. 6 The abundance of 3 Cas proteins in the 2 phyla (Proteobacteria and Firmicutes) is probably due to a higher number of sequenced genomes for these corresponding phyla present in the NCBI database. A previous study also discovered a similar bias in the number of sequenced genomes being from these 4 phyla. 46 The above findings demonstrate that Cas3, Cas9, and Cas10 proteins do not exhibit similar distributions among all major bacterial phyla; however, the distribution biases are not of the inherent characteristics of the respective phyla, instead the members of available complete genome sequences in the database (Figure 2).
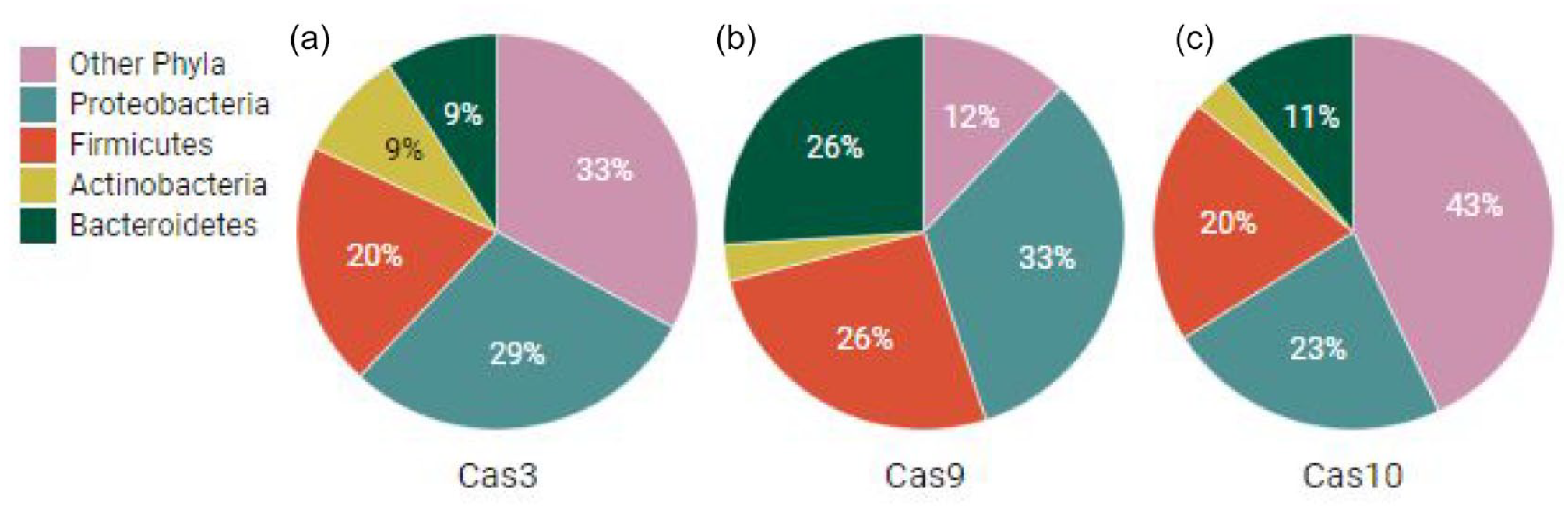
Distribution of (a) Cas3, (b) Cas9, and (c) Cas10 across bacterial phyla.
Evolutionary relationships of Cas proteins
Across all 3 Cas protein families, both within and between the family members, there is a low to moderate amount of amino acid sequence conservation ranging from 30% to 50% amino acid identity. An unrooted phylogenetic tree as shown in Figure 3 exhibits the evolutionary relatedness of Cas-associated proteins of different bacterial species that possess those corresponding reference proteins.

The phylogenetic tree of Cas3, Cas9, and Cas10 proteins.
Cas proteins were separated into 3 distinct clades, Cas3, Cas9, and Cas10, which revealed that these 3 proteins originated monophyletically. It was observed that the branch length of the clades representing Cas3 and Cas10 proteins is relatively longer than that of clades representing Cas9 proteins (results not shown in the tree), suggesting that the members of the Cas3 and Cas10 proteins evolved more rapidly than those of the members of Cas9 protein family. It is interesting to note that all clades representing Cas9 and Cas10 proteins diverged from a common clade of Cas3 proteins and later diverged into Cas9 and Cas10 protein families. The phylogenetic tree also reveals that Cas3 proteins represent the higher number of clades compared with the number of clades represented within Cas9 or Cas10 proteins. This finding is attributed maybe due to a large number of species representing Cas3 proteins included for the tree construction. Overall, the branch lengths and number of branches within the Cas3 family suggests that members of the Cas3 family evolved and diverged more rapidly than the members of the Cas9 or Cas10 protein families. Although, the phylogenetic tree of the Cas proteins was not reported earlier, a study suggests a common ancestry of the effector complexes of type I and type III Cas systems in which Cas3 and Cas10 belongs, respectively. 6
Protein domains experience differential selection pressures
Neutral theory of molecular evolution accounts for the occurrence of both non-synonymous and synonymous substitutions. 47 The ratio between the 2 types of substitutions has been used to determine the strength of structure-function constraints. 48
Specifically, the ratio (ie, dN/dS and denoted by ω) of the non-synonymous substitution rate (dN) to the synonymous substitution rate (dS) indicates the evolution of the structure-function constraint of the different domains or the whole protein. A dN/dS value is smaller than 1 means that the protein can accumulate more synonymous (silent) substitutions than non-synonymous (missense) substitutions. A dN/dS value approximately equal to 1 means that the mutations have no negative or positive effects on an organism’s ability to survive and reproduce.47,48 In addition, neutral mutations alter amino acids without changing their chemical properties. Such neutral mutations can be fixed or fluctuate in a population. Finally, a dN/dS value greater than 1 indicates that the protein accumulates more missense mutations, which change amino acids with different chemical properties. Such mutations result in new functions under positive selection.
The rate of non-synonymous changes (dN), the rate of synonymous changes (dS), and the ratio of the non-synonymous to the synonymous changes (dN/dS) for the whole protein (corresponding to the first error bar in each subplot) and different protein domains (corresponding the remaining error bars in each subplot) for Cas3, Cas9, and Cas10 proteins are shown in Figure 4a to c, respectively. As shown in Figure 4a, Cas3 proteins have the 5 domains: an HD-nuclease domain, 2 Super Family 2 (SF2) helicase domains, a linker domain, and a C-terminal domain (CTD). Overall, Cas3 proteins experiences negative selection pressure, but different protein domains are functionally constrained differently. Of the 5 domains, the HD-nuclease domain and the 2 SF2 helicase domains (RECA1 and RECA2) appear to be experiencing negative selection, and they are highly conserved. This finding supports a previous observation that the helicase domain of Cas3 is highly conserved among the members of the Cas3 protein family. 49 The SF2 helicase domain is an ATPase responsible for unwinding the target DNA strand. This ATPase activity allows the HD-nuclease domain to cleave the ssDNA strand of the target sequence. 13 The importance of these domains for the destruction of foreign DNA makes it obvious that these domains are under negative selective pressure. Conversely, the CTD shows a dN/dS value above 1, which indicates the positive selection pressure in that region and the low conservation. The CTD appears to be responsible for interacting with Csel, which is a protein in the CASCADE array that functions to recognize the PAM sequence in target DNA. 50 The rapid evolution of this domain has likely occurred to accommodate the high degree of variation in PAM sequence recognition across bacterial species.
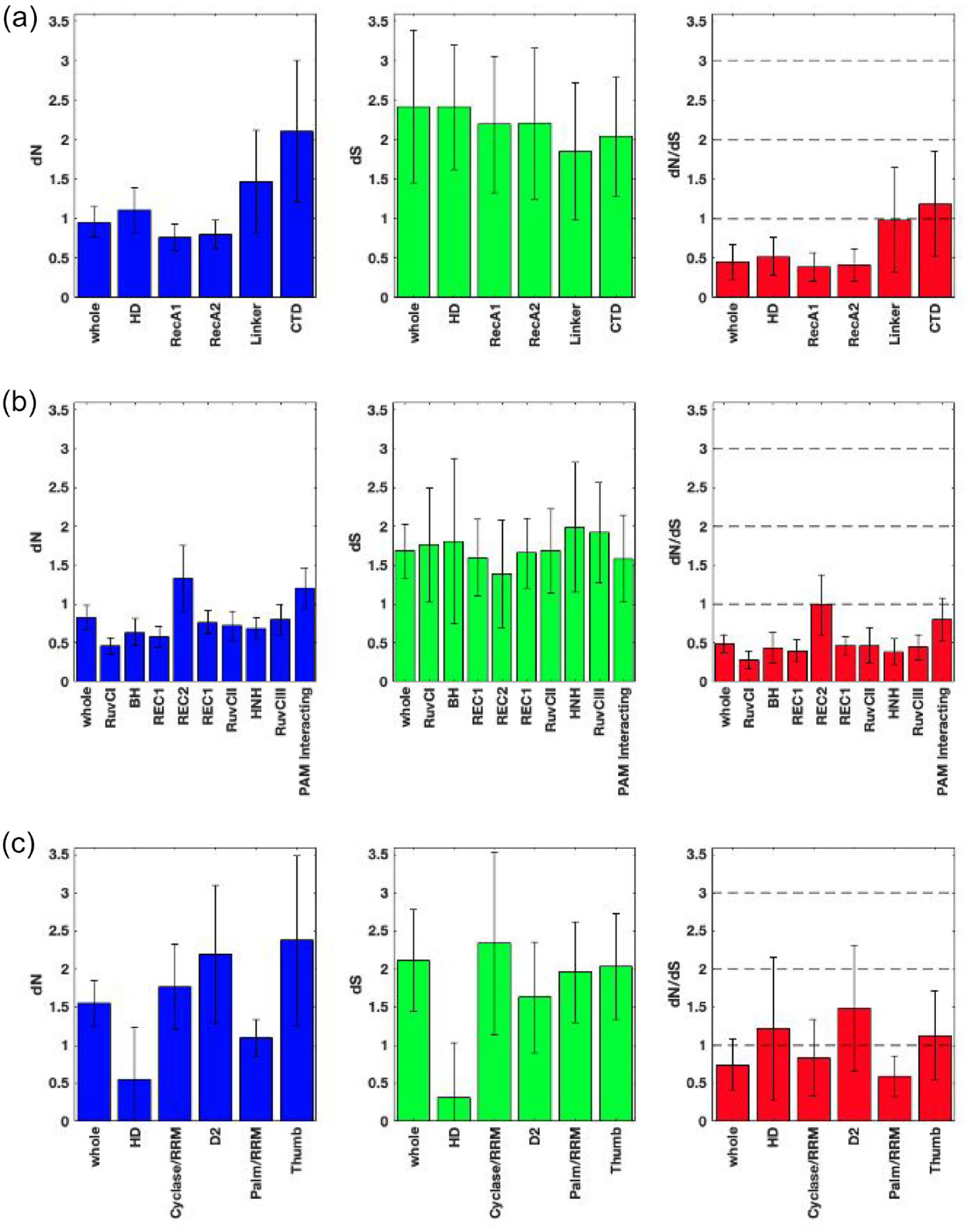
The rate of non-synonymous substitutions (dN), the rate of synonymous substitutions (dS), and the dN/dS ratio for (a) Cas3, (b) Cas9, and (c) Cas10. The structure of Thermofibida fusca (T fusca) Cas3 consists of 5 domains, including HD (residues 1-258), RecA1 (residues 259-545), RecA2 (residues 546-777), Linker (residues 778-834), and CTD (residues 834-944). Through a pair-wise alignment with T fusca, the domain positions of Escherichia coli (E coli) Cas3 are obtained as follows: HD (residues 1-267), RecA1 (residues 259-545), RecA2 (residues 546-777), linker (residues 778-834), and CTD (residues 834-944).
As shown in Figure 4b, the structure-function constraint analysis for Cas9 proteins has similar conservation patterns to those seen for Cas3 proteins. Overall, Cas9 proteins experience negative selection. It has 9 different domains, where 7 domains (including 2 REC1/RECI, bridge helix (BH), HNH, and 3 RuvCs) maintain high levels of sequence conservation, experience negative selective pressure with dN/dS less than 0.5, and correspond to the functions such as binding, cleaving, and destructing the target DNA. 51 As shown in Figure 4, whole protein sequences in each Cas family are highly constrained; thus, they are experiencing negative selection, where Cas3 and Cas9 are relatively more constrained than Cas10. All these domains are crucial to the functionality of the whole system, so a higher level of constraint (dN/dS < 1) is expected. The PAM-interacting (PI) domain of Cas9 appears to be experiencing negative selection. As previously discussed for Cas3, a higher level of mutations is expected to allow the level of variations in the PAM sequences; however, Cas9 PAM seems different and shows less sequence variation.
The REC1/RECI domain which remains uncharacterized also demonstrated a dN/dS value above 1, indicating that it has experienced positive selective pressure. A similar finding, stating that the REC lobe is one of the least conserved regions across all members of the Cas9 family, was also reported in a previous study. 51 This further suggests that a high level of conservation of the REC2/RECII domain might not be required for the functionality of the system.
As shown in Figure 4c, Cas10 proteins have different 5 domains including HD nuclease, Fingers, Zn Finger/motif, Palm/RRM and Thumb. Although Cas10 proteins overall experience negative selection as seen in the case of Cas3 and Cas9 proteins, their protein domains show evolutionary constraints with dN/dS values close to 1 or above 1, which are relatively higher number than dN/dS values for most domains in both Cas3 and Cas9. It indicates that Cas10 domains experience a lower level of sequence conservation as well as less structure-function constraint. A previous study similarly demonstrated that 85% of the Cas10 protein family have conserved polymerase active-site motifs; however, only 36% of the Cas10 protein family have conserved HD-nuclease domain. 52 Therefore, it can be suggested that the Cas10 protein is experiencing positive selection, and its various domains evolved more rapidly than the domains of Cas3 and Cas9 protein families. As the function of the Cas10 protein remains poorly understood, it is difficult to correlate their functions with differences of structure-function constraints. This further corroborates the phylogenetic relationship that these proteins have evolved rapidly and show little similarity in functionality across protein families.
Streptococcus pyogenes Cas9 is organized with recognition (REC) and nuclease (NUC) lobes. The REC lobe consists of a long alpha-helix called BH and multiple alpha-helical REC domains (REC1 [residues 94-179], REC2 [residues 180-307], REC1 [residues 308-717]), whereas the nuclease (NUC) domain comprises 2 endonuclease domains (HNH [residues 775-908]) and 3 RuvCs (RuvCI [residues 1-59], RuvCII [residues 718-774], and RuvCIII [residues 909-1098]) and a PI domain (residues 1099-1368).
Pyrococcus furiosus Cmr2 (PDF ID 4W8Y) Cas10 comprises HD domain (residues 1-216), Cyclase/RRM (residues 216-503) including Zn finger domain at the end, D2 (residues 503-593) whose residues are based on Zhu and Ye 20 and Manav et al 21 but whose function is unclear, Palm/RRM (residues 593-763) domain, and Thumb (residues 764-871) domain.
Conclusion
Based on the results of protein conservation, phylogenetic tree, and structure-function analyses, the following conclusions are made: CRISPR-associated Cas3, Cas9, and Cas10 proteins are prevalent in bacteria and archaea, and they are distributed among major bacterial phyla. All 3 proteins have their monophyletic origins with Cas9 and Casl0 first being commonly diverged from Cas3, and the 2 were later separated into separate protein families. The analysis of the branch lengths and the number of clades suggest that members of the Cas3 family evolved more rapidly than the members of the Cas9 or Cas10 protein families. Cas3 and Cas9 proteins, including their most of functionally interactive domains, experience negative selection, whereas Cas10 protein along with its all functionally interactive domains experience positive selection. The result supports that Cas10 protein and its functionally interactive domains may have evolved new molecular functions that yet to be characterized.
Future work includes similar analyses of other proteins associated with type I, II and III systems that would provide further information about the origins and functions of the CRISPR systems. Also, structure-function constraints of the different functionally interactive domains may be further exploring the scope of an alternate and improved genome editing system to the currently used CRISPR-Cas9 system. In addition, the utilization of the CRISPR-Cas3/Cas10 may provide additional clues to the recently discovered immunity to S pyogenes Cas9 and Staphylococcus aureus Cas9 systems. 53
Footnotes
Acknowledgements
The authors acknowledge the contribution of Dorcie Gillette for conducting the data analysis, and the financial support for her thesis project from the Honors College, Sam Houston State University.
Author Contributions
Weerakkody Ranasinghe contributed for the manuscript preparation and revision. Dorcie Gillette contributed for the initial data collection and analysis. Alexis Ho contributed for the manuscript preparation and proof reading. Hyuk Cho contributed for the study design, data analysis, manuscript preparation and revision. Madhusudan Choudhary contributed for the study conception, study design, manuscript preparation and revision.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.