
Abstract
Background:
Clostridium botulinum and Clostridium perfringens, 2 major foodborne pathogenic fusobacteria, have a variety of virulent protein types with nervous and enterotoxic pathogenic potential, respectively.
Objective:
The relationship between the molecular evolution of the 2 Clostridium genomes and virulence proteins was studied via a bioinformatics prediction method. The genetic stability, main features of gene coding and structural characteristics of virulence proteins were compared and analyzed to reveal the phylogenetic characteristics, diversity, and distribution of virulence factors of foodborne Clostridium strains.
Methods:
The phylogenetic analysis was performed via composition vector and average nucleotide identity based methods. Evolutionary distances of virulence genes relative to those of housekeeping genes were calculated via multilocus sequence analysis. Bioinformatics software and tools were used to predict and compare the main functional features of genes encoding virulence proteins, and the structures of virulence proteins were predicted and analyzed through homology modeling and a deep learning algorithm.
Results:
According to the diversity of toxins, genome evolution tended to cluster based on the protein-coding virulence genes. The evolutionary transfer distances of virulence genes relative to those of housekeeping genes in C. botulinum strains were greater than those in C. perfringens strains, and BoNTs and alpha toxin proteins were located extracellularly. The BoNTs have highly similar structures, but BoNT/A/B and BoNT/E/F have significantly different conformations. The beta2 toxin monomer structure is similar to but simpler than the alpha toxin monomer structure, which has 2 mobile loops in the N-terminal domain. The C-terminal domain of the CPE trimer forms a “claudin-binding pocket” shape, which suggests biological relevance, such as in pore formation.
Conclusions:
According to the genotype of protein-coding virulence genes, the evolution of Clostridium showed a clustering trend. The genetic stability, functional and structural characteristics of foodborne Clostridium virulence proteins reveal the taxonomy and diverse distribution of virulence factors.
Introduction
Clostridium is a diverse genus (more than 100 species) of gram-positive, endospore-bearing obligate anaerobes that are widespread in the environment. 1 Clostridium botulinum (C. botulinum) and Clostridium perfringens (C. perfringens), which are among the principal foodborne pathogens, cause toxin-mediated disease either via preformed toxins or through the formation of toxins in the enteric tract. 2 Strains of C. botulinum are traditionally classified into 7 types, A through G, based on their botulinum neurotoxin type (BoNT/A through BoNT/G), with types A, B, E, and F causing most cases of botulism in humans, 3 whereas types C and D are responsible for botulism in animals. 4 C. perfringens also has 7 toxin types (A-G), which are based on combinations of the 6 main toxin proteins (Alpha, Beta, Epsilon, Iota, CPE, and NetB). Types A and C are associated with various diseases in humans, such as enterotoxemia, antibiotic-associated and sporadic diarrhea, and food poisoning 5 ; types B, D, E, and F cause diseases in various animals. 6 In C. botulinum, typical BoNTs are composed of an N-terminal 50 kDa light chain and a C-terminal 100 kDa heavy chain linked by a disulfide bond and are solely responsible for botulism, leading to flaccid paralysis and death.7,8 All C. perfringens isolates produce alpha-toxin, a zinc-containing phospholipase C enzyme of 370 amino acids that consists of a membrane-binding C-domain and a catalytic N-domain composed of α-helices and a β-sheet. 9 Type C isolates produce beta1 or beta2 toxins, which are the protoxins of 35- and 28-kDa β-pore-forming toxins, respectively, and belong to the α-hemolysin family. 10 The CPE protein, a 35 kDa single polypeptide, consists of an N-terminal cytotoxicity domain and a C-terminal receptor-binding domain that mediates membrane insertion during pore formation and oligomerization.11,12
Previously, the foodborne pathogen C. botulinum was divided into 4 phylogenetic groups according to a 16S rRNA gene sequence analysis. 4 C. botulinum was subsequently expanded to include groups V and VI because of the discovery of new toxin strains.13,14 Further research identified additional clusters within these groups, indicating genetic diversity: 5 larger clusters in group I, 8 different gene clusters in group II, and 3 different clusters in group III.15 -17 Studies have shown that C. perfringens can carry many toxin-producing genes. Owing to the wide distribution of C. perfringens, asymptomatic carriage by healthy people and the migration of many virulence gene plasmids have led to the formation of the unique virulence gene of C. perfringens. 18 Different sources and hosts of C. perfringens show significant genetic diversity and unique epidemiological molecular characteristics. 19 Human food poisoning-related C. perfringens strains are clustered into one large cluster and are scattered in several other clusters in the evolutionary tree. Interestingly, isolates within different clusters were found to express the same type of toxin; conversely, those in a single cluster could express different types of toxins. 17 This discordant phylogeny between the host bacteria and the toxins indicates that horizontal transfer of the genes encoding the toxins is responsible for the variation observed within the species. 13 Many studies have examined the evolutionary classification of Clostridium virulence genes, but few comparative analyses of virulence gene evolution and the relative evolutionary distance or of virulence protein structural and functional prediction in foodborne Clostridium are available. 20
In this work, the genome sequences, virulence gene sequences, and predicted virulence proteins of thirty-eight foodborne strains of C. botulinum and thirty-four foodborne strains of C. perfringens were comparatively analyzed. This research aimed to examine the phylogenetic relationship between the main foodborne Clostridium strains and their virulence factors to comparatively analyze the relative genetic distance of C. botulinum and C. perfringens virulence genes and to analyze the functional and structural characteristics of their virulence proteins.
Methods
The procedures used for this study are illustrated in Figure 1.

Schematic representation of the procedures used in this study.
Screening and evaluation of genomic and virulence protein-encoding gene sequences
Thirty-eight C. botulinum strains and thirty-four C. perfringens strains that infect humans, have annotation information integrity, and have complete assemblies were selected for a comparative analysis from the NCBI database; the detailed information on the samples is shown in Supplemental Table 1 and Table 2. The datasets that passed the completeness and contamination tests consisted of sequences submitted before January 11, 2024. For the evolutionary analysis, Clostridium scatologenes, which has a defined phylogenetic and taxonomic status (ATCC 25775) and is closely related to C. botulinum and C. perfringens in Clostridium, was used as a control strain. As shown in Supplemental Table 3, the length, G + C content, and accession numbers of the strains were downloaded from the NCBI database. The contamination and completeness of the genomic sequences were evaluated with the KBase server (https://www.kbase.us/) via CheckM software v1.0.18. 21
Phylogenetic analyses of the strain genomes
Based on the whole-genome amino acid sequences of each strain, we used the CVTree4 webserver (http://cvtree.online/v4/prok/index.html) to construct the first phylogenetic tree. This method uses composition vectors without sequence alignment and the parameter setting K-tuple 6. 22 Each genome sequence is represented by a composition vector that uses the Markov model to calculate the difference between the prediction frequencies and the k-string frequencies. 23 The tree was modified via MEGA-X v10.2.2. 24 The same data were analyzed for average nucleotide identity (ANI) via the JSpecies webserver (https://jspecies.ribohost.com/jspeciesws/) to verify the significance of the first phylogenetic tree. 25 ANI dendrograms, which were generated from Newick format files via Njplot, 26 were constructed via the distance matrix from the distance value (DV). The formula DV = 1-(ANI value), which was balanced by the average value method, was calculated with the DrawGram tool of the PHYLIP package (v3.695), as described by Baum. 27
Multilocus sequence analysis (MLSA) of virulence genes
The sequences of 38 C. botulinum strains and thirty-four C. perfringens strains downloaded from the NCBI database were searched for housekeeping and virulence genes. We selected the housekeeping genes DNA topoisomerase (ATP-hydrolyzing) subunit B (gyrB), recombinase RecA (recA), pyrroline-5-carboxylate reductase (proC), phosphate acetyltransferase (pta), translation elongation factor 4 (lepA), glutamine-hydrolyzing GMP synthase (guaA), and F0F1 ATP synthase subunit beta (atpD) for the C. botulinum strains and DNA topoisomerase (ATP-hydrolyzing) subunit B (gyrB), recombinase RecA (recA), glycerol kinase GlpK (glpK), guanylate kinase (gmk), collagenase ColA (colA), chaperonin GroEL (groL), and quinolinate synthase NadA (nadA) for the C. perfringens strains to calculate the basic DVs of the species. These housekeeping genes were scattered throughout the chromosome, and the gene sequences within the operons of all of the strains were conserved, did not require gene rearrangement, and were suitable for MLSA. The housekeeping genes were the same length in all of the strains, and the different virulence genes were the same length. The different groups were divided by the types of virulence genes (bont/a, bont/b, bont/e, and bont/f in the C. botulinum strains; alpha, beta-2, and cpe in the C. perfringens strains), and we concatenated the base sequences for subsequent MLSA. We used the MEGA-X tool to calculate the distances of the concatenated genes via the NJ algorithm with 100 bootstrap replicates, which is based on the discrete gamma distribution of the Tamura–Nei model. 24 The model applied to calculate the DVs in the MLSA was an ideal alternative model based on the function of “find best DNA/protein models.” 28 The same settings were used to calculate all phylogenetic DVs to ensure the comparability of the results. We calculated the relative changes in the average number of inherited DVs among the virulence genes as the DVs of concatenated housekeeping genes minus the DVs of individual housekeeping genes, which represents the change in the number of inherited DVs during virulence gene transfer.
Prediction and comparative analysis of virulence protein-encoding genes and structural characteristics
The SMART tool was used to predict the domain architecture of the virulence proteins (http://smart.embl-heidelberg.de/). 29 PSORTb software (http://www.psort.org/psortb) was employed to predict the subcellular locations of the virulence proteins. 30 SIGNALP-5.0 (http://www.cbs.dtu.dk/services/SignalP) and TMHMM (http://www.cbs.dtu.dk/services/TMHMM) were used to predict signal peptide cleavage sites and transmembrane helices.31,32 The SWISS-MODEL server (http://swissmodel.expasy.org) and AlphaFold software v2.1.160 (https://github.com/deepmind/AlphaFold) were used to predict the three-dimensional (3D) structures of the toxin proteins.33 -35 We transformed the amino acid sequences of the virulence proteins into the FASTA format and generated 3D virulence protein structures via homology modeling. Structural alignment and editing were performed using PyMOL Molecular Graphics System v2.0 (http://www.pymol.org).
Results
General genomic characteristics and sequence quality assessment
A summary of the features of the thirty-eight C. botulinum genomes, thirty-four C. perfringens genomes, and the control genome (C. scatologenes) is provided in Supplemental Table 3. The genome sizes of the C. botulinum and C. perfringens strains varied from 3.66 to 4.43 Mb (standard deviation, SD = 0.18 Mb), with N50/L50 values ranging from 3.66 to 4.26 Mb/scaffold, from 2.94 to 3.60 Mb (SD = 0.18 Mb), and from 2.90 to 3.44 Mb/scaffold, respectively (as shown in Supplemental Table 3). The G + C contents of the thirty-eight C. botulinum genomes and thirty-four C. perfringens genomes ranged from 27.37% to 28.30% (SD = 0.25%) and 28.12% to 28.50% (SD = 0.10%), respectively. Compared with those of the control genome (C. scatologenes, 5.75 Mb and 29.60%), the genomes of C. botulinum and C. perfringens were much smaller and presented lower G + C contents. The contamination levels of the C. botulinum and C. perfringens sequences were 0% to 1.38% and 0% to 0.44%, respectively, and the completeness values of the sequences were 98.06% to 100.00% and 98.39% to 100.00%, respectively (as shown in Supplemental Table 4). These results revealed that these sequences had high quality, with high completeness (values > 98.06%, the acceptable level is >95%), and low contamination levels (values < 1.38%, the acceptable level is <5%). The potential toxicity risk based on the presence of the virulence genes of 38 C. botulinum strains and thirty-four C. perfringens strains identified in this study are listed in Supplemental Table 4. The toxin types, which were determined by analyzing the virulence genes, were type A (encoded by bont/a, 20/38), type B (encoded by bont/b, 7/38), type E (encoded by bont/e, 1/38), type F (encoded by bont/f, 6/38), type A + B (encoded by bont/a and bont/b, 3/38), and type A + F (encoded by bont/a and bont/f, 1/38) in the C. botulinum strains and type A (encoded by alpha, 8/34), type C (encoded by alpha + beta-2, 5/34), type F (encoded by alpha + cpe, 11/34), and type C + CPE (encoded by alpha + beta-2 + cpe, 10/34) in the C. perfringens strains (as shown in Supplemental Table 4).
Phylogenetic composition vector (CV) and average nucleotide identity (ANI) analyzes
Phylogenetic analyses using both composition vector (CV)- and average nucleotide identity (ANI)-based methods were conducted on the 38 C. botulinum strains and 34 C. perfringens strains, with C. scatologenes as the outgroup species. The CV method utilizes whole-genome amino acid sequences to construct a phylogenetic tree (Figure 2). In this tree, C. botulinum and C. perfringens strains formed distinct groups, labeled parts A and B, respectively. Part A had 6 clusters dominated by type A or A+ (A1, A3, and A5) and type B, E, or F (A2, A4, and A6) toxin risk regions, although strains such as F634 and ATCC 3502 were exceptions. Part B included 5 clusters dominated by type A or C + CPE (B1, B2, and B5) and type A or C (B3 and B4) regions, with outliers such as MGYG-HGUT-02372, 149/92 and CPI 75-1.

Results of the CV analysis of the phylogenetic tree of 38 C. botulinum strains, 34 C. perfringens strains, and the C. scatologenes control strain used in this study. A, B, C, E, F, and CPE indicate the toxin types.
ANI analyses were performed for each of the 2 bacterial populations to verify these findings separately and refine the genetic relationships, resulting in 2 additional dendrograms (Figures 3 and 4). In the second dendrogram, the regional correspondence with the phylogenetic tree (Figure 2) was as follows: regions (a1, a2, and a4) and (a3 and a5) were the same as regions (A1, A3, and A5) and (A2, A4, and A6), respectively. In the third dendrogram, regions (b1, b2, and b3) and (b4 and b5) were the same as regions (B1, B2, and B5) and (B3 and B4), respectively. The 2 dendrograms corroborated the clusters observed in the CV phylogenetic tree, with new minor shifts noted for strains such as ATCC 3502 and CPN 17a across different evolutionary clusters. Notably, the regions identified in the ANI analysis mirrored those identified via the CV method, highlighting the consistency of the genetic groupings based on the toxin type.
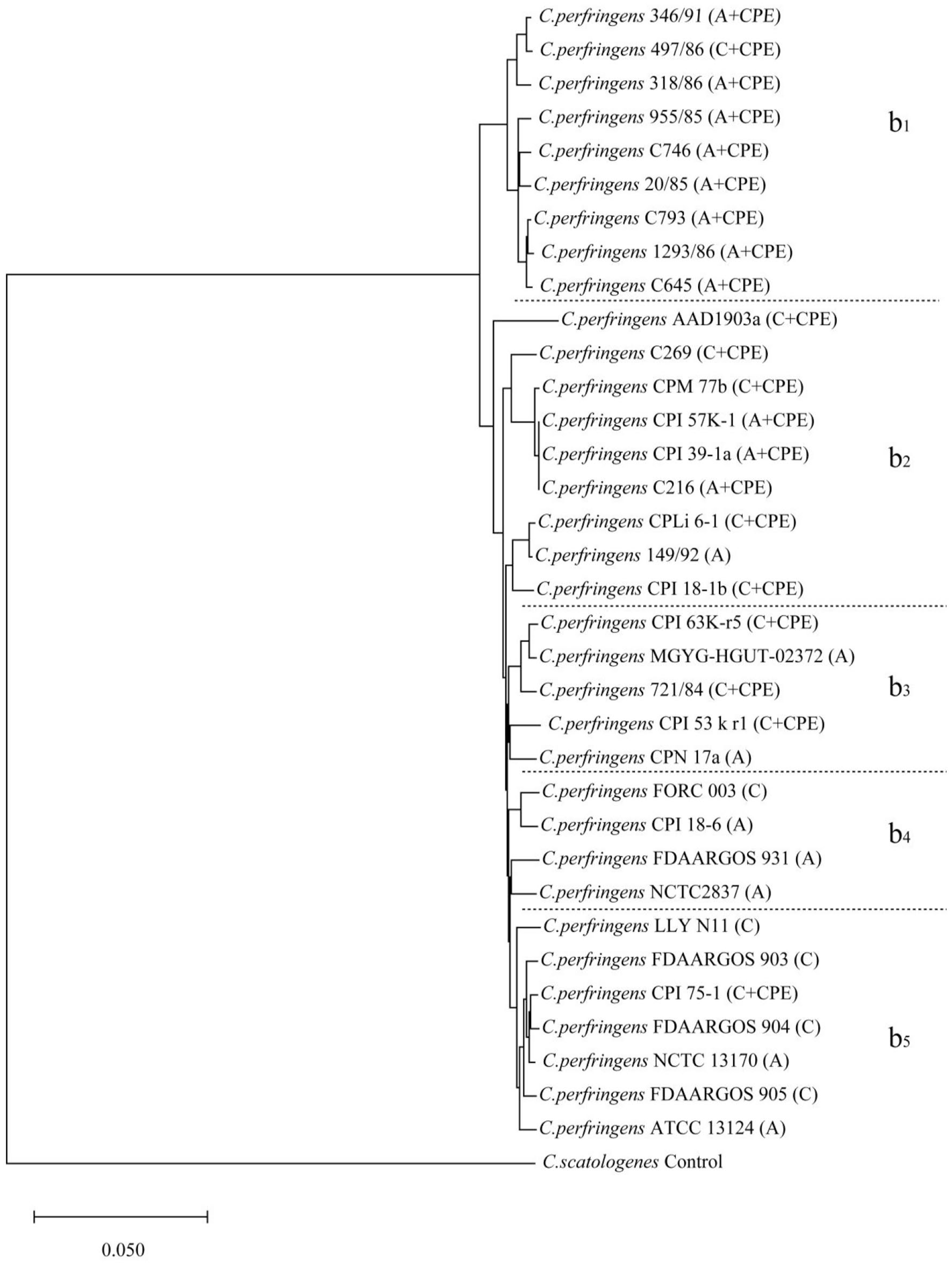
Results of the ANI analysis of the dendrogram of 38 C. botulinum strains and one control strain. A, B, E, and F indicate the toxin types.
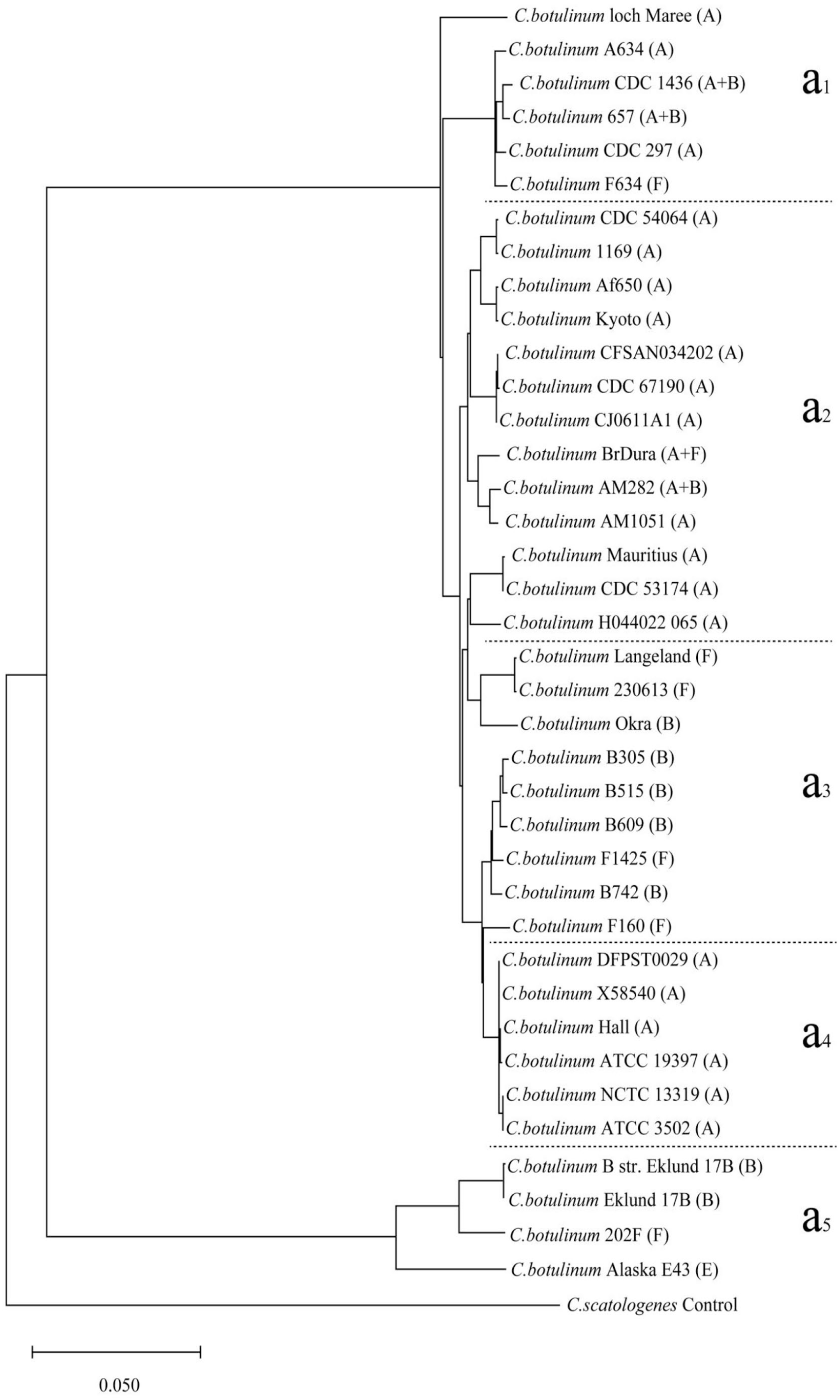
Results of the ANI analysis of the dendrogram of 34 C. perfringens strains and one control strain. A, C, and CPE indicate the toxin type.
Phylogenetic distance analysis
We analyzed the phylogenetic relationships and evolution of virulence gene transfer by comparing the bont/a, bont/b, bont/e, bont/f, alpha, beta-2, and cpe genes of the seventy-two strains with the housekeeping genes of the strains. The sequences of 7 housekeeping proteins (GyrB-RecA-ProC-Pta-LepA-GuaA-AtpD and gyrB-RecA-GlpK-GmK-ColA-GroEL-NadA) were concatenated from the genomes of the C. botulinum and C. perfringens strains, respectively. Because the distance values (DVs) of the relative genetic drift distance of virulence genes between the control strains (ATCC25775) and the representative strains of C. perfringens (ATCC13124) and C. botulinum (ATCC3502) were equal (shown in Supplemental Table 5), we evaluated the relative degree of virulence gene transfer by calculating the mean differences between the ATCC3502 and ATCC13124 strains and the thirty-seven and thirty-three other strains of C. botulinum and C. perfringens, respectively. The types and numbers of virulence genes (shown in Supplemental Table 4) were as follows: toxin type A (20/38), type B (7/38), type E (1/38), type F (6/38), type A + B (3/38), and type A + F (1/38) in the C. botulinum strains and toxin type A (8/34), type C (5/34), type F (11/34), and type C + CPE (10/34) in the C. perfringens strains. The relative DVs, from high to low, were as follows: 0.1751 (type F), 0.1704 (type B), 0.1353 (type E), 0.0619 (type A), 0.0560 (type A + F), and 0.0287 (type A + B) in the C. botulinum strains and 0.0130 (type A), 0.0126 (type C + CPE), 0.0105 (type C) and 0.0101 (type F) in the C. perfringens strains (shown in Figure 5 and Supplemental Table 5). The DVs of the virulence genes in the C. botulinum strains were greater than those of the virulence genes in the C. perfringens strains. The DVs of single virulence genes (types A, B, E, or F) were greater than those of double virulence genes (types A + B or A + F) in the C. botulinum strains, which was not observed in the C. perfringens strains. The average evolutionary DVs of types B and F were greater than the DVs of types E and A among the single virulence genes; the DVs of types A + F were greater than those of types A + B among the double virulence genes in the C. botulinum strains. All of the average evolutionary DVs in the C. perfringens strains showed relatively little change.
Average different DVs of the ATCC3502 strain compared with those of 37 other strains of C. botulinum and the ATCC13124 strain compared with those of 33 other strains of C. perfringens for virulence genes + housekeeping genes and housekeeping genes, as analyzed by MLSA.
Relevant predictive indicators of virulence protein localization and functional domains
We obtained the subcellular localization prediction scores of the 7 virulence proteins (shown in Table 1). BoNT/A, BoNT/B, BoNT/E, BoNT/F, and alpha-toxin were extracellular (all with scores of 9.98). The localization of beta2 toxin and enterotoxin could not be identified because their scores were only 3.33 and 2.50, respectively. The prediction results for all of the virulence proteins revealed no transmembrane regions or helices. The cleavage sites of alpha- and beta2-toxin were predicted to be at positions 28 to 29 and 30 to 31, with probability values of .9725 and .9502, respectively. As shown in Table 1 and Figure 6, the 4 domains of BoNT/A, BoNT/B, BoNT/E, and BoNT/F were peptidase_M27 (ranging from 2 to 418), toxin_trans (ranging from 518 to 866), toxin_R_bind_N (ranging from 859 to 1079), and toxin_R_bind_C (ranging from 1063 to 1293). The domains of alpha-toxin were ZN_dep_PLPC (ranging from 1 to 278) and PLAT (ranging from 286 to 398), and the domain of enterotoxin was Clenterotox (ranging from 54 to 240), whereas beta2-toxin had none.
Predicted subcellular localization, signal peptide, transmembrane region, helix number, cleavage site, and domains of the virulence proteins of representative C. botulinum and C. perfringens strains.

-, none.

The domain start and end sites and cleavage sites were predicted by SMART and SignalP software with the sequences of the virulence genes of the strains.
Prediction results and evaluation of scores for virulence protein structures
Phylogenetic trees constructed from the virulence gene sequences of BoNT/A, BoNT/B, BoNT/E, BoNT/F, alpha-toxin, beta2-toxin and CPE (shown in Figure 7) revealed that the genetic relationships between BoNT/A and BoNT/B, between BoNT/E and BoNT/F, and between alpha-toxin and beta2-toxin were closer than those of the other components, and CPE had the weakest correlation.

Phylogenetic tree of the sequences of the virulence proteins of the strains.
These genetic relationships are further reflected in the 3D toxin protein structure templates summarized in Table 2. Notably, BoNT/E is the closest template to BoNT/F, with an acceptable sequence identity of 62.18%. The SWISS-MODEL server revealed high sequence identities for the templates of BoNT/A (99.92%), BoNT/B (95.43%), BoNT/E (98.08%), alpha-toxin (99.73%), and CPE (96.81%). The global model quality estimates (GMQEs) for these models range between 0.76 and 0.87, exceeding the threshold of 0.5, and their sequence coverages are between 0.88 and 1.00, surpassing the 0.85 benchmark. These metrics indicate the reliability of the model constructs, with one exception: the beta2 toxin model has a low GMQE of 0.14 and a sequence coverage of only 0.32. A reliability score was assigned to each residue from 0 to 1, indicating the expected similarity to the native structure. The higher the value is, the greater the reliability of the residue. 36 Generally, more than 30% of the sequence identity for each template was acceptable, but the closest template for beta2 toxin was an uncharacterized protein (with a low sequence identity of 10.47% and a GMQE evaluation value of 0.14). 37
Prediction parameters and results for the toxin protein structures obtained via AlphaFold software and the SWISS-MODEL tool with the sequences of the same virulence proteins from the strains. -: none; GMQE: global model quality estimate. plDDt: predicted local-distance difference test, ipTM + pTM: multimer prediction evaluation.

AlphaFold software was employed for secondary structure predictions to validate these findings, particularly the beta2 toxin model, and to obtain structural comparisons and the CPE trimer structure. The predicted local-distance difference test (plDDt) values for the monomers ranged from 86.53 to 92.44, indicating a high prediction accuracy. Additionally, the CPE trimer score of 0.27 (ipTM + pTM) provides an insight into its structural stability.
Comparison of the structural characteristics of virulence proteins in C. Botulinum
Owing to the sequence similarity of the respective templates (as shown in Table 2), we established homology models based on the respective structures. BoNT/A, BoNT/B, BoNT/E, and BoNT/F had highly similar structures, and all of the structures had 3 main domains, namely, a “binding” domain, a “translocation” domain, and a “catalytic” domain, in C. botulinum (shown in Figures 8A-D and 9A-F). The “binding” domain, which consists mainly of β-strands connected by a prominent α-helix, appeared as 2 different subdomains of roughly equal size. The N-terminal subdomain had 2 seven-stranded β-sheets sandwiched together to form a jelly roll pattern, whereas the C-terminal subdomain had a similar size and adopted a modified β-trilobal fold to form a seven-stranded β-barrel structure next to the N-terminal jelly roll pattern. The main body of the “translocation” domain had a cylindrical shape, in which the helices and antiparallel strand were twisted around each other, similar to a spiral coil. At either end of the pair of helices, a shorter α-helix was arranged parallel to the axis of the long helix. In addition, the domain had 2 chain-like parts that were arranged parallel to the α-helices, forming loops that were disordered or nicked in BoNT/A and BoNT/B (Figures 8A, D, and 9A-C) but were intact in BoNT/E and BoNT/F (Figures 8B, C, and 9D-F). The “catalytic” domain was a mixture of α-helix and β-chain secondary structures whose active sites were deeply buried in the protein and accessible via a channel. 38 The 3 functional domains of BoNT/E and BoNT/F were similar to the 3 corresponding functional domains of BoNT/A and BoNT/B. Although the individual domains were similar, the conformational arrangements of the domains were different. In BoNT/A and BoNT/B, the “catalytic” and “binding” domains were located on either side of the long translocation domain, and no interaction was observed between them (Figures 8A, D, 9B, and C), but the 2 domains interacted on the same side of the translocation domain in BoNT/E and BoNT/F (Figures 8B, C, 9E, and F). Structural comparisons also confirmed these results (Figure 9A and D).

Results of the SWISS-MODEL prediction. (A–D, F–H) Structures of BoNT/A, BoNT/E, BoNT/F, BoNT/B, alpha-toxin, beta2-toxin, and CPE. The structures in A to D are annotated with “catalytic,” “translocation,” and “binding” domains; the structures in b and c are annotated with disulfide loops. The structures in E and F are annotated with the N- and C-terminal domains; the structure in e is annotated with loops 1 and 2; and the structure in g is annotated with the “binding receptor” and “beta-pore-forming” structures.

Results of the AlphaFold prediction. A and D Superposition of the structures of BoNT/A (green) and BoNT/B (white) and of the structures of BoNT/E (purple) and BoNT/F (yellow), respectively. B, C, E, and F Structures of the BoNT/A, BoNT/B, BoNT/E, and BoNT/F monomers, respectively, which are annotated with the “catalytic,” “translocation,” and “binding” domains. H Structure of the CPE monomer, which is annotated with the “binding receptor” and “beta-pore-forming” structures. I and J Structures of alpha-toxin and beta2-toxin, annotated with the “N-terminal domain” and “C-terminal domain,” respectively. G The structure of the CPE trimer, which is annotated with the “claudin-binding pocket.” In addition, the structures in D, E, F, and I are annotated with “loop” structures.
Comparison of the structural characteristics of virulence proteins in C. Perfringens
In C. perfringens, the structure of the alpha-toxin protein contains 2 domains connected by 2 loops. The 2 domains were a six-helical N-terminal domain containing the active site and a six-stranded β-sheet sandwiched C-terminal domain, which was previously implicated in membrane binding (Figures 8E and 9I). 39 The structure of beta2 toxin (Figure 8F), which forms a multimeric functional complex with 2 α-helical N-terminal domains and a three-β-sheet sandwiched within the C-terminal domain, is similar to that of the alpha-toxin protein. We detected multiple β-tongue strands in beta2 toxin (Figure 9J). However, they are not visible in Figure 8F because of the prediction method used, possibly because of the low sequence identity in the homology model (as shown in Table 2). The monomer of the CPE had an elongated shape (Figures 8G and 9H) composed of seventeen-stranded β-sheets and 5 helices. The “binding receptor” domain formed nine-stranded β-sheets sandwiched with a short helical element. The “beta-pore-forming” (five β-sheets, three α-helices, and six β-sheets) domains formed a module that exhibited an elongated caterpillar shape. The C-terminal domain of the CPE trimer formed a “claudin-binding pocket” shape, which was entirely in one plane on the same side of the molecule (Figure 9G).
Discussion
In this study, a molecular evolutionary analysis of the whole-genome amino acid sequences of 38 C. botulinum strains and 34 C. perfringens strains was performed. In terms of the potential toxicity risk, the strains evolved into distinct clusters, which were dominated by type (A, A+) or type (B, E, F) regions in C. botulinum and type (A, C) or type (F or C + CPE) regions in C. perfringens. Both phylogenetic methods revealed that the virulence genes of C. botulinum and C. perfringens tended to cluster. These results suggest that virulence gene transfer among Clostridium strains may have occurred during the evolution of virulence, which may be related to the frequent exchange of pathogenic factors.13,40 In this work, we observed that the virulence genes were dominated by bont/a, bont/a + (bont/b or bont/f), bont/b, bont/e, and bont/f in C. botulinum and cpa, cpa+ + cpb2 or cpa + cpe, and cpa + cpb2 + cpe in C. perfringens. These toxin genes of foodborne Clostridium strains are located on plasmids (enterotoxin genes on chromosomes or plasmids), except for the alpha-toxin gene, which is chromosomally located. Related toxin genes have been identified in different species of Clostridium, and the variation in some toxin genes indicates the horizontal transfer of toxin genes and subsequent independent evolution from strain to strain. 41 Owing to the prevalence of horizontal gene transfer in bacterial ecosystems, increasing numbers of “core” genes, such as housekeeping and virulence genes, is thought to play a central role in shaping the patterns of nucleotide substitution and polymorphism based on the refined species classification. 42 However, the discordant evolutionary distributions of individual virulence genes could have been caused by other factors, and the unknown mechanism needs to be further investigated.
BoNT/A, BoNT/B, BoNT/E, and BoNT/F (encoded by bont/a, bont/b, bont/e, and bont/f, respectively) and alpha-toxin, beta2-toxin, and CPE (encoded by cpa, cpb2, and cpe) represent the significant toxins produced by C. botulinum and C. perfringens among foodborne Clostridium strains, respectively. These toxins can cause food poisoning in humans and are involved in severe diseases. 43 Studies have shown that toxigenic Clostridium species are not all phylogenetically related and that the genetic stability of virulence genes is different. 44 The MLSA results indicated that the DVs of virulence genes in C. botulinum strains were greater than those of virulence genes in C. perfringens strains. The results suggested that the genetic stability of the virulence genes of C. perfringens strains was greater than that of C. botulinum among foodborne pathogenic Clostridium strains. All toxin-producing C. perfringens strains carry the alpha-toxin gene, which is located at the same site on a variable region of the chromosome close to the origin of replication, improving the overall stability of the C. perfringens strains, and other toxin genes are located on plasmids. 41 The genetic stability of the double virulence genes was lower than that of the single virulence genes in C. botulinum, which may be the reason for the high cytotoxic activity and stable inheritance of the single virulence protein of C. botulinum; however, this phenomenon was not observed in the C. perfringens strains because of the impact of the vertical inheritance of the cpa gene. In C. botulinum, the inheritance of the type F virulence gene is the most unstable, and the type F toxin is indeed a particularly variable neurotoxin. 45 However, owing to the different selection methods used to calculate molecular evolution distances, the lack of pangenomic global analysis and the lack of consideration of the negative regulatory mechanisms of antibiotic resistance genes, these results may have several limitations.
At present, the virulence of foodborne pathogens is generally accepted to not be entirely driven by toxin genes because the expression of toxin genes is highly complex and may be influenced by the strain specificity of transcription, translation, and posttranslational modifications.46 -48 The results of this study revealed that the locations of all the virulence proteins were extracellular, and none of these toxins were present in the cytoplasm or in the membrane or other organelles. Therefore, some other functional proteins may regulate and assist in the transport of cytotoxins across the membrane to accomplish their toxic effects. Some complex regulatory mechanisms are involved in the production and transport of virulence genes, which lead to the expression of different genetic traits of Clostridium virulence factors. The current classification of Clostridium species hinders functional and trait predictions, and the diversity and number of toxins produced by some strains, as well as the surprising potency of some of the toxins produced by these pathogens, are not fully understood. 49
We accurately predicted the 4 similar functional domains of the amino acid sequences of the BoNT/A, B, E, and F toxins, which have different starting and ending sites. The peptidase_M27 domain, which includes the zinc protease, functions by blocking neurotransmitter release though cleavage of the syntaxin, synaptobrevin, and SNAP-25 proteins. 50 The toxin_trans domain is a central translocation domain with a unique long pair of helical structures, and the toxin_R_bind_(N or C) domain is an N- or C-terminal receptor-binding domain. 51 The sequences of alpha-toxin and beta2-toxin contained cleavage sites and N-terminal signal peptides, indicating that the toxin is secreted via the secretory translocation pathway. The 2 domains of alpha-toxin are Zn_dep_PLPC, which has zinc-dependent phospholipase C activity, 52 and PLAT, which binds to a variety of membrane- or lipid-associated proteins. 53 CPE is the single Clenterotox domain encoded by enterotoxin, 54 and beta2 has no domain according to our domain prediction. This result indicates the limitations of predictive methods and the need for further research.
The 7 serotypes (A–G) of C. botulinum neurotoxins have significant sequence homology and similar structure–function relationships. 55 BoNT/A, BoNT/B, BoNT/E, and BoNT/F are three-component toxin structures (Figures 8A-D and 9A-F), which are composed of catalytic, translocation, and binding domains.56 -58 The active site of the “catalytic” domain is buried deep in the protein and has a negatively charged surface, which is critical for docking with the substrate. The “translocation” domain is thought to allow the endosome to penetrate and form a pore, allowing the “catalytic” domain to cross the membrane into the cytoplasm. The “binding” domain can bind the presynaptic nerve ending, which is the first step in the intoxication mechanism. 51 In BoNT/E, the “catalytic” domain and the “binding” domain are on the same side as the “translocation” domain, and all 3 have a common interface 58 ; we obtained the same structural arrangement in the results for the predicted BoNT/F structure. This unique association may influence the rate of translocation, which may account for the faster rate of toxicity. In addition, the disordered or nicked disulfide loops in BoNT/A and BoNT/B are more exposed and intact than those in BoNT/E and BoNT/F and may play important and unique roles in translocation. The presence of different subsets of the same structural motifs in various toxins may indicate an evolutionary mechanism in which stable functional domains are assembled into modular units that produce virulence. 51
The alpha-toxin, beta2-toxin, and CPE (Figures 8E-G and 9H-J) monomers form polymers to perform their toxic functions. We obtained the same alpha-toxin structure with the C-terminal and N-terminal domains using 2 prediction models; the N-terminal domain is the “catalytic” domain with an active site and substrate binding function, and the C-terminal domains can bind calcium ions and interact with other proteins. 59 Additionally, 2 mobile loops are present in the N-terminal conformational domain that open and close the active site and play crucial roles in toxicity.39,60 We obtained a satisfactory prediction model of the beta2 toxin structure (Figure 9J) via the AlphaFold prediction method. Beta2 toxin is a functional oligomeric pore-forming toxin, and its receptors are located mainly in the lipid rafts of HL-60 cells, where oligomers form and induce cytotoxic effects. 61 CPE is a major cause of food poisoning and antibiotic-associated diarrhea. 62 The “receptor binding” domain consists of the C-terminal region of CPE, whereas the “beta-pore-forming” domain consists of the N-terminal region, which is structurally homologous to the family of aerolysin-like β pore-forming proteins.63,64 The predicted compact trimer structure (Figure 9G) of CPE has been verified in several initial structures, especially the claudin-binding pocket site, suggesting that it may have some biological relevance, such as in pore formation.
Conclusions
In this work, we described the molecular evolution and functional and structural diversity of the virulence factors of foodborne C. botulinum and C. perfringens strains. Phylogenetic analyses revealed that the C. botulinum and C. perfringens strains tended to cluster based on the virulence genes. The genetic stability of the virulence genes of the C. perfringens strains was greater than that of the virulence genes of the C. botulinum strains among the foodborne pathogenic Clostridium strains, and the presence of a single virulence gene had greater relative genetic stability than the double gene in C. botulinum. All of these virulence proteins need other functional proteins to regulate and assist in transporting cytotoxins across the membrane to achieve toxic effects. The spatial arrangements and disulfide loop structures of BoNT/E and BoNT/F differ from those of BoNT/A and BoNT/B, and the 2 mobile loop structures of alpha-toxin and the claudin-binding pocket of CPE may play important and unique roles in their functions. Overall, the results of this study provide useful information for further understanding the taxonomical and diverse distributions of virulence factors of foodborne Clostridium strains.
Supplemental Material
sj-docx-1-evb-10.1177_11769343241294153 – Supplemental material for Comparative Phylogenetic Analysis and Protein Prediction Reveal the Taxonomy and Diverse Distribution of Virulence Factors in Foodborne Clostridium Strains
Supplemental material, sj-docx-1-evb-10.1177_11769343241294153 for Comparative Phylogenetic Analysis and Protein Prediction Reveal the Taxonomy and Diverse Distribution of Virulence Factors in Foodborne Clostridium Strains by Ming Zhang and Zhenzhen Yin in Evolutionary Bioinformatics
Footnotes
Acknowledgements
We acknowledge the contributions of Dr. Hengzhi Wu and Bin Cheng to the Biomedical Big Data Center, Nanfang College, Guangzhou.
Author Contributions
Conceived and designed the experiments: Ming Zhang. Analyzed the data: Zhenzhen Yin. Wrote the first draft of the manuscript: Ming Zhang. Contributed to the writing of the manuscript: Zhenzhen Yin. Approved the results and conclusions described in the manuscript: Zhenzhen Yin. Jointly developed the structure and arguments for the paper: Zhenzhen Yin. Made critical revisions and approved the final version: Ming Zhang. All of the authors reviewed and approved the final manuscript.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Guangdong Province University Characteristic Innovation Project (2023KTSCX202) and the Guangdong Province Education Science Planning Project (Higher Education Special Project) (2024GXJK460).
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.