
Abstract
Pseudomonas aeruginosa is a major cause of nosocomial infections and is often associated with biofilm-mediated antibiotic resistance. The LasR protein is a key component of the quorum system in P. aeruginosa, allowing it to regulate its biofilm-induced pathogenicity. When the bacterial population reaches a sufficient density, the accumulation of N-(3-oxododecanoyl) acyl homoserine lactone (3O-C12-HSL) leads to the activation of the LasR receptor, which then acts as a transcriptional activator of target genes involved in biofilm formation and virulence, thereby increasing the bacteria’s antibiotic resistance and enhancing its virulence. In this study, we performed a structure-based virtual screening of a natural food database of 10 997 compounds against the crystal structure of the ligand-binding domain of the LasR receptor (PDB ID: 3IX4). This allowed us to identify four molecules, namely ZINC000001580795, ZINC000014819517, ZINC000014708292, and ZINC000004098719, that exhibited a favorable binding mode and docking scores greater than −13 kcal/mol. Furthermore, the molecular dynamics simulation showed that these four molecules formed stable complexes with LasR during the 150-ns molecular dynamics (MD) simulation, indicating their potential for use as inhibitors of the LasR receptor in P. aeruginosa. However, further experimental validation is needed to confirm their activity.
Introduction
Pseudomonas aeruginosa (P. aeruginosa) is a Gram-negative bacterium responsible for a variety of acute and chronic infections worldwide. It is a major contributor to almost all hospital infections, primarily among patients with burn wounds, severe leukemia, urinary tract, and bloodstream infections, and plays a significant role in targeting immunocompromised patients with cystic fibrosis, neutropenic cancer, or infected by human immunodeficiency virus (HIV). 1 - 4 Thus, the World Health Organization (WHO) has placed P. aeruginosa at the top of the list of critical pathogens for which new antibiotics are urgently required. 5
The biofilm mode of growth of P. aeruginosa and expression of virulence factors, such as pyocyanin, pyoveridine, rhamnolipid, exopolysaccharide, and elastase, reportedly make it more resistant to a large number of important antibiotics, thereby increase its pathogenicity. 6 - 9 This ability of biofilm formation induces 20% to 30% resistance to fluoroquinolones, including ciprofloxacin and levofloxacin, as well as 13% to 22% to gentamicin.10,11 The control or inhibition of virulence and biofilm mode of growth may be a promising therapeutic strategy to reduce the pathogenicity of infections caused by P. aeruginosa and improve or boost the efficiency of the available antibiotics. 12 - 14
The biofilm production in P. aeruginosa is governed by a mechanism called quorum sensing (QS).8,9 The QS is a cell-to-cell communication mechanism that implies producing, releasing, sensing, and responding to extracellular signaling molecules known as autoinducers (AIs) which control and regulate the collective behavior of bacteria. 15
P. aeruginosa has four interconnected hierarchies of QS namely las, iqs, pqs, and rhl, with a significant dominance for las (LasR-LasI) and rhl (RhlR-RhlI) systems which regulate the majority of physiological and virulence factors. 16 These two pathways use two AI molecules, namely N-(3-oxododecanoyl) acyl homoserine lactone (3O-C12-HSL) and N-butanoyl acyl homoserine lactone (C4-HSL) that interact with their cognate receptors LasR and RhlR, respectively. 17 The las system is considered to be at the top of the QS hierarchy where the activation of LasR can trigger the cascade which subsequently activates other pathways and regulates the expression of a set of genes responsible for virulence and biofilm production. 18
Given its position in the P. aeruginosa QS hierarchy, the LasR receptor has become a potential target for anti-microbial therapy. 19 There is a considerable amount of literature describing inhibitors for LasR from the past 15 years, and these can be classified into four categories: AHL-like antagonists, non-AHL-like antagonists, covalent binders, and natural-product-based inhibitors. 20 However, to the best of our knowledge, no candidate has further been pushed into clinical trials or approved by the Food and Drug Administration.
The current study aimed to identify new active food compounds targeting the ligand-binding pocket of the LasR receptor of P. aeruginosa. In this regard, we performed a virtual screening (VS) of a natural food compounds database which contains 10 997 purchasable food constituents against the ligand-binding domain LasR crystal structure (PDB code: 3IX4), followed by molecular dynamics (MD) simulations for the four top-scoring hits.
Material and Methods
Protein structure preparation
The crystal structure of the ligand-binding domain of the LasR receptor was retrieved from the PDB database (PDB ID: 3IX4) 21 and prepared in AutoDockTools 22 by removal of water and solvent molecules, removal of the bound TP-1 ligand, addition of polar hydrogens, and partial charge assignment.
Structure-based virtual screening
The structure-based VS was performed at the MTi-OpenScreen webserver (https://bioserv.rpbs.univ-paris-diderot.fr/services/MTiOpenScreen/) against the natural food compound database (which contains 10 997 purchasable food constituents). 23 The MTi-OpenScreen integrates AutoDock Vina tools and provides five electronic drug-like chemical libraries, and among them, the food database contains suitable compounds for docking and purchasability according to the Zinc15 database after physicochemical and toxicophore filtration. 23 The grid box was created around the TP-1 binding site with a grid center of x, y, and z dimensions of 1.5, 14.92, and 5.86, respectively, and its size has been set to 20 × 20 × 20 Å. The results of the VS were ranked according to the binding energy of their best scoring conformation. The top-ranked molecules as well as the TP-1 were then subjected to re-docking using an installed AutoDock Vina. The docking results were visualized and analyzed using Pymol, with the ligand interaction diagram implemented in Maestro and PLIP web tool. 24
Molecular dynamics
MD simulation was used to study the stability of the top-four-ranked hits in complex with LasR over time. MD was carried out for 150 ns with a recording interval of 75 ps using Desmond module. In the system builder, the OPLS3e force field was selected, TIP3 P was used as a solvent model with a 10 Å orthorhombic box, and then the system charge was neutralized by adding 0.15 M of sodium (Na+) and chloride ions (Cl−). The generated model system was subjected to energy minimization and equilibrated via an NPT ensemble at a constant temperature of 310 K and 1.01325 bar pressure. All other Desmond parameters were kept at their default values. Once MD simulation was done, simulation trajectories were analyzed using the simulation interaction diagram included in the Desmond module. The principal component analysis (PCA) was performed using the Bio3D package implemented in R. 25
In silico prediction of pharmacokinetic properties
The SwissADME webserver was used for computing the physicochemical, pharmacokinetic, and druglikeness properties of top-scoring compounds (ZINC000001580795, ZINC000014819517, ZINC000014708292, and ZINC000004098719). 26 The canonical simplified molecular-input line-entry system (SMILES) of these compounds was used and submitted to SwissADME webserver.
Results and Discussion
Structure-based virtual screening analysis
The identification and screening of natural food compounds against a particular disease target can offer a promising avenue for the discovery of potent inhibitors with improved binding affinity and reduced toxicity. These compounds can potentially serve as lead ligands in drug discovery, providing a foundation for the development of effective therapeutics. To streamline the research process and minimize unnecessary experiments, VS has emerged as a valuable computational technique. By employing VS, researchers can efficiently identify a focused set of small molecules that exhibit significant affinity for the active pocket of the targeted receptor.
In the current study, we performed VS against a food library (contains 10 997 purchasable food constituents), which is implemented in MTi-OpenScreen webserver in order to identify new hits targeting the LasR receptor, therefore inhibiting the production of P. aeruginosa virulence factors. First, to validate our used VS protocol, we re-docked TP-1 (2,4-Dibromo-6-({[(2-nitrophenyl)carbonyl]amino}methyl)phenyl 2-chlorobenzoate) against the crystal structure of the LasR receptor (PDB ID: 3IX4). TP-1 is a triphenyl (TP) scaffold-based compound that has the potential to activate the LasR receptor, even though it does not have significant chemical similarities to the 3O-C12-HSL. In fact, TP-1 and its derivatives TP-2, TP-3, and TP-4 were found to directly activate LasR with binding affinities that are several orders of magnitude lower than any other class of inhibitors. In addition, TP-1 and its derivatives do not cross-react with related signaling receptors such as QscR, LuxR, and RhlR. In this study, TP-1 was used as the reference molecule, and its docking score as the threshold value.
As a result, we found that the re-docked TP-1 has a similar conformation and occupies a similar position within the ligand-binding pocket as compared to the bound TP-1 from the original structure (root mean square deviation [RMSD] was significantly lower with a value of 1.8 Å) (Figure 1). It should be noted that for a prediction of ligand-target conformations, an RMSD cutoff value less than 2 Å is considered good. 27 The VS results revealed that one molecule with a binding affinity score of −14.5 kcal/mol, 3 molecules with −13 kcal/mol, 18 molecules with −12 kcal/mol, 65 molecules with −11 kcal/mol, and 50 molecules with −10.5 kcal/mol (Supplemental Table S1). Among our identified hits, some molecules have already been found to be potential inhibitors, such as naringenin (e.g. ZINC000000001785 [−11.6 kcal/mol]), 28 benzamide compounds (e.g. ZINC000012343956 [−10.5 kcal/mol]), 29 resveratrol compounds (e.g. ZINC000004098633 (−10.8 kcal/mol), ZINC000108555676 (−10.8 kcal/mol), and ZINC000253615309 (−10.6 kcal/mol).19,30
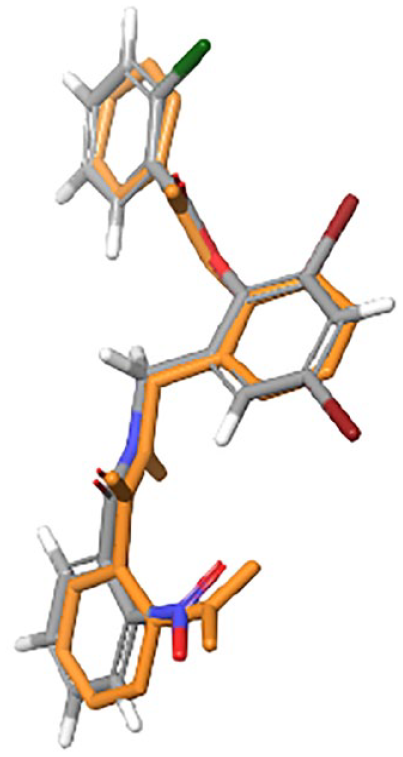
Superposition of crystallographic (gray) and re-docked (orange) triphenyl (TP)-1.
In this study, we focused only on molecules with a binding affinity score of −14 and −13 kcal/mol, given that the re-docking of TP-1 (used as a reference) showed a binding affinity of −14.2 kcal/mol (Table 1). These molecules are 3,3’-Dihydroxy-4,5-dimethoxybibenzyl (ZINC000001580795), Lupiwighteone (ZINC000014819517), Shinpterocarpin (ZINC000014708292), and R_Glabridin (ZINC000004098719).
2-D Interaction diagram, interaction summary and binding energy of TP-1, ZINC000001580795, ZINC000014819517, ZINC000014708292, and ZINC000004098719.

In the 2-D interaction diagram, residues in green and blue colors represent the hydrophobic and polar interactions, respectively. Pink arrows show the hydrogen bond formation, and green lines indicate Pi-Pi stacking interactions.
Abbreviation: TP, triphenyl.
To the best of our knowledge, none of these molecules have previously been identified as inhibitors of LasR, nor have they demonstrated any activity against P. aeruginosa. However, these molecules have shown other potential medicinal activities. The R_Glabridin, an isoflavonoid isolated from the roots of Glycyrrhiza glabra L, possesses diverse potential properties including antioxidant, anti-inflammatory, estrogenic, and antiproliferative properties in human breast cancer cells, neuroprotective effects, as well as its application in skin-whitening and anti-obesity effects. 31 - 33 It was reported as well that this molecule exhibited considerable anti-staphylococcal activity against various clinical isolates of methicillin-resistant strain of Staphylococcus aureus with a minimum inhibitory concentration of 12.5 μg ml−1. 34 Lupiwighteone, a prenylated flavonoid isolated from various Lupinus plants, has potential activities such as anti-inflammatory and anticancer properties, 35 and studies have revealed that lupiwighteone induces apoptotic cell death in breast cancer by effectively inhibiting the PI3 K/Akt/mTOR pathway. 36 Moreover, in prostate cancer cells (DU-145) and neuroblastoma cells (SH-SY5Y), lupiwighteone exhibited cytotoxic, apoptotic, and antiangiogenic activities. 37 Beyond its anticancer properties, lupiwighteone has also demonstrated antifungal activities against Phytophthora infestans. 38 Shinpterocarpin, an isoflavonoid compound extracted from the root of Glycyrrhiza glabra L. (Leguminosae), the stem bark of Erythrina sacleuxii, and the root bark of Erythrina abyssinica 39 showed moderate antitumor effects against various tested cancer lines with IC50 values spanning from 15 to 53 μM. 40 In addition, 3,3’-dihydroxy-4,5-dimethoxybibenzyl derived from Dendrobium williamsonii has exhibited cytotoxic effects against KB and MCF-7 cancer cells. 41
The analysis of the co-crystallized agonist (TP-1) revealed that it interacts mainly with LasR through hydrogen bonds with residues Tyr 56, Trp 60, Asp 73, and Ser 129 (Table 1 and Figure 2). On the other hand, the docking mode of the four studied molecules showed that ZINC000001580795 interacts through H-bond with Tyr 56 and Ser 129, ZINC000014819517 interacts through H-bond with Trp 60 and Leu 110, and ZINC000014708292 interacts through H-bond with Leu 110 while ZINC000004098719 has not made any H-bond interaction and interacted mainly through Pi-Pi stacking interaction (Table 1 and Figure 2). The difference in the interaction modes between TP-1, which is an agonist molecule, and the identified molecules may induce a variation in the modulation of the LasR receptor by generating an antagonistic conformation.

The potential binding poses for TP-1, ZINC000004098719, ZINC000001580795, ZINC000014708292, and ZINC000014819517 in complex with LasR. TP indicates triphenyl.
Molecular dynamic analysis
Molecular docking can predict and provide insight into the interaction mode between the ligand and the protein in a static state. To validate the predicted interaction mode of the four hits under dynamic conditions and to monitor the conformations changes of the receptor induced by these ligands, we performed 150 ns of MD simulation. The MD results were analyzed using different parameters such as (1) protein RMSD that measures the conformational changes of a given complex over time and describes whether the simulation is in equilibrium 42 (Figure 3A), (2) protein root mean square fluctuation (RMSF) that characterizes local changes along the protein chain (Figure 3B), (3) PCA analysis that determines the total motions of the Cα atoms in the protein indicated by eigenvectors of the covariance matrix to investigate the complexes’ stability (Figure 4), and (4) protein–ligand contact analysis that presents the fraction of the active residues implicated in the ligand interaction 43 (Figure 5).

Protein RMSD (A) and protein RMSF (B) analyses of C-alpha atoms of TP-1, ZINC000001580795, ZINC000014708292, ZINC000004098719, and ZINC000014819517 in complex with LasR. The letter H indicates alpha-helix, and β indicates beta-sheet. RMSD indicates root mean square deviation; RMSF, root mean square fluctuation; TP, triphenyl.

PCA of TP-1 (A), ZINC000001580795 (B), ZINC000014819517 (C), ZINC000014708292 (D), and ZINC000004098719 (E) in complex with LasR. PCA results which include graphs of PC2 vs PC1, PC2 vs PC3, and PC3 vs PC1 colored from blue to red in order of time (the blue specifies initial timestep, white specifies intermediate, and the final timestep is represented by red color), and an eigenvalue rank plot (scree plot). In the eigenvalue plot, the cumulative variance is labeled for each data point. PCA indicates principal component analysis; TP, triphenyl.

Protein–ligand contact monitoring of triphenyl (TP)-1 (A), ZINC000001580795 (B), ZINC000014819517 (C), ZINC000014708292 (D), and ZINC000004098719 (E) in complex with LasR. The left histograms show interaction fraction with active amino acid residues. The x-axis presents the residues involved in the interactions, and the y-axis presents the normalized value of the temporal length of the interactions during the simulation. The stacked bar charts are normalized over the course of the trajectory: For example, a value of 0.7 suggests that 70% of the simulation time, the specific interaction is maintained. Values over 1 indicate that the protein residue could make multiple contacts of the same subtype with the ligand. The middle schematic representation (2D simulation interaction diagram) of ligands indicates the percentage of interactions with the protein residues. The right plot indicates the timeline of protein–ligand contacts during the simulation for the five complexes. Some residues made more than one specific contact with the ligand, which is represented by a darker shade of orange, according to the scale to the right of the plot.
The RMSD analysis of C-alpha atoms showed that the complexes of LasR with ZINC000014819517 (1.80 ± 0.30 Å), ZINC000004098719 (1.85 ± 0.14 Å), and ZINC000001580795 (1.69 ± 0.18 Å) have a similar RMSD value compared to that of the LasR-TP-1 complex (1.65 ± 0.21 Å), indicating that these molecules form stable complexes with LasR throughout the simulation. The RMSD values were higher in LasR-ZINC000014708292 (2.89 ± 0.41 Å) complex, indicating that ZINC000014708292 may induce significant conformational changes within the LasR structure upon binding. Further examination of the RMSF plots revealed effectively that the binding of ZINC000014708292 induces significant fluctuations across all the LasR structures with an average value of 1.54 ± 0.64 Å. The same effects but with a lesser extent were observed in the RMSF of LasR-ZINC000014819517 (1.12 ± 0.61 Å) which induced localized conformational fluctuations in the Loops connecting H1 H2, β1-β2, and β3-β4. On the other hand, the complexes of LasR with ZINC000001580795 (0.92 ± 0.40 Å) and ZINC000004098719 (0.82 ± 0.44 Å) showed lower overall RMSF fluctuations than that of TP-1 (0.95 ± 0.40 Å).
The PCA results align consistently with the RMSF results. By analyzing the first three principal components (PC1, PC2, and PC3) that account for the most variance in the original data (i.e. the largest uncorrelated motion in the trajectory) 44 of tp1 and the four other systems, we observed that ZINC000014708292 accounted for higher variances of 56.2% for the total of the first three PCs, indicating that this ligand increased the motion of the protein’s Cα-atoms, 45 followed by ZINC000014819517, which showed a variance of 48.7%. ZINC000001580795 and ZINC000004098719 exhibited a variance of 36.1% and 36.7%, respectively, meaning that these two ligands induced more stable movement of the protein’s Cα atoms than TP1, which showed a variance of 33.9% for the total of the first three PCs.
Monitoring of protein–ligand contacts throughout the simulation showed that each molecule exhibits a different interaction mode with the LasR ligand-binding pocket compared to TP-1 (Figure 4). Despite this difference in terms of residues and type of interactions involved (eg, hydrogen bonds, hydrophobic, ionic, and water bridge), all ligands were maintained stable within the LasR binding pocket through a network of favorable and persistent interactions. This may indicate that each ligand may modulate the LasR activity differently through specific interactions which are dependent on the chemical structure of each molecule, as each one has a different scaffold.
In silico prediction of pharmacokinetic properties
The molecular, physicochemical, pharmacokinetic, and druglikeness properties of the four selected compounds were predicted using SwissADME 26 and summarized in Table 2. In general, these compounds did not violate the druglikeness properties, specifically Lipinski et al, 46 Ghose et al, 47 Veber et al, 48 Muegge et al, 49 and Egan et al 50 rules, and have characteristics similar to those of oral drugs with an oral bioavailability probability of 0.55%.
Physicochemical, pharmacokinetics, and druglikeness properties of ZINC000001580795, ZINC000014819517, ZINC000014708292, and ZINC000004098719.

Abbreviations: BBB, blood–brain barrier; GI, gastrointestinal; Log Kp, skin permeability; TPSA, topological polar surface area in Å.
All compounds had optimal lipophilicity (MLogP < 5) and showed adequate values in terms of their molecular weight (<500 g/mol) and polar surface area (20 < TPSA < 130 Å), which is essential for good penetration through biological membranes. Furthermore, all compounds were within the normal range of the number of hydrogen bond acceptors (⩽10), bond donors (⩽5), and rotatable bonds (⩽10) according to Lipinski’s rule of five and Veber’s rule. 26
All four analyzed compounds showed high gastrointestinal (GI) absorption and good skin permeation (log Kp). Except for ZINC000014819517, all compounds exhibited blood–brain barrier (BBB) permeability, indicating that remaining three compounds are able to cross into the brain.
Conclusion
The current study attempts to identify new LasR antagonists from the food compounds database to inhibit biofilm production and consequently attenuate P. aeruginosa virulence and antibiotic resistance. By combining structure-based VS and MD simulation approaches, we were able to identify four molecules, namely ZINC000004098719, ZINC000001580795, ZINC000014708292, and ZINC000014819517, that have a favorable binding affinity to LasR and exhibit a significant stability throughout 150 ns of the MD simulation. This computational study provides confidence that these four molecules can be proposed as potential biofilm-lowering candidates targeting the LasR receptor. Further experimental validations are needed, such as assessing the impact of these molecules on biofilm formation, quantifying pyocyanin and rhamnolipid, and performing competition binding assays between 3O-C12-HSL and the identified ligands for their binding to LasR, as well as analyzing mRNA expression of genes regulated by lasR (ie, rhl and pqs genes). Once experimentally and clinically validated, these molecules could potentially serve as dietary supplements in the case of infections caused by P. aeruginosa to boost the efficiency of the clinically used antibiotics.
Supplemental Material
sj-xlsx-1-bbi-10.1177_11779322231212755 – Supplemental material for In Silico Identification of Natural Food Compounds as Potential Quorum-Sensing Inhibitors Targeting the LasR Receptor of Pseudomonas aeruginosa
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322231212755 for In Silico Identification of Natural Food Compounds as Potential Quorum-Sensing Inhibitors Targeting the LasR Receptor of Pseudomonas aeruginosa by Meryam Magri, El Mehdi Bouricha, Mohammed Hakmi, Rachid EL Jaoudi, Lahcen Belyamani and Azeddine Ibrahimi in Bioinformatics and Biology Insights
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out under National Funding from the Moroccan Ministry of Higher Education and Scientific Research to AI. This work was also supported by a grant to AI from the Institute of Cancer Research and the PPR-1 program to AI.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.